

Key Points
MCD-associated genetic lesions strongly promoted accumulation of B cells in splenic GCs of unimmunized mice
Mice engineered to harbor 4 distinct MCD-associated genetic lesions showed a massive expansion of splenic GCs and developed DLBCL upon aging.

Abstract
Unique molecular vulnerabilities have been identified in the aggressive MCD/C5 genetic subclass of diffuse large B-cell lymphoma (DLBCL). However, the premalignant cell-of-origin exhibiting MCD-like dependencies remains elusive. In this study, we examined animals carrying up to 4 hallmark genetic lesions found in MCD consisting of gain-of-function mutations in Myd88 and Cd79b, loss of Prdm1, and overexpression of BCL2. We discovered that expression of combinations of these alleles in vivo promoted a cell-intrinsic accumulation of B cells in spontaneous splenic germinal centers (GCs). As with MCD, these premalignant B cells were enriched for B-cell receptors (BCRs) with evidence of self-reactivity, displayed a de novo dependence on Tlr9, and were more sensitive to inhibition of Bruton’s tyrosine kinase. Mutant spontaneous splenic GC B cells (GCB) showed increased proliferation and IRF4 expression. Mice carrying all 4 genetic lesions showed a >50-fold expansion of spontaneous splenic GCs exhibiting aberrant histologic features with a dark zone immunophenotype and went on to develop DLBCL in the spleen with age. Thus, by combining multiple hallmark genetic alterations associated with MCD, our study identifies aberrant spontaneous splenic GCBs as a likely cell-of-origin for this aggressive genetic subtype of lymphoma.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is an aggressive heterogeneous clinical entity that is thought to be derived from germinal center (GC) B cells or cells that have recently passed through the GC. Subtypes of DLBCL have been identified based on differences in gene expression or enrichment for certain genetic alterations that carry distinct outcomes to therapy.1-4 The activated B cell–like (ABC) gene expression subtype of DLCBL carries a poor prognosis after immunochemotherapy.1 The MCD/C5 genetic class is a subset of ABC-DLBCL enriched for gain-of-function mutations in MYD88 and CD79B.2-4 Mutant MYD88 and CD79B are essential for the survival of MCD-DLBCL cell lines, where they allow for formation of a super-complex containing MYD88, Toll-like receptor 9 (TLR9), and the immunoglobulin M (IgM) B-cell receptor (BCR) (My-T-BCR) that promotes oncogenic signaling via NF-κB.5 This complex can be abolished by therapies targeting BCR-mediated activation of NF-κB through inhibition of Bruton’s tyrosine kinase by molecules such as ibrutinib or through depletion of TLR9. Importantly, MCD-DLBCL is highly enriched for self-reactive IgM BCRs, and survival of MCD-DLBCL cell lines is dependent on this self-reactivity.6
GCs are necessary for antibody affinity maturation that is critical for humoral immunity and are deeply associated with the development of lymphomas.7 Although GCs are most often studied after immunization or infection, they can form in a variety of tissues at homeostasis. For example, GCs can form spontaneously in the spleen where they accumulate with age and may contribute to autoimmunity.8 Unlike GCs in other settings, accumulation of spontaneous splenic GCs is promoted by Myd88 and Tlr7 and suppressed by Tlr9.9
Memory B cells (MBCs) and antibody-producing plasma cells (PCs) are the principal cellular outputs from the GC. Although it has been proposed that MCD-DLBCL arises from plasmablasts (PBs) that are in the process of exiting the GC reaction, this has not been formally shown in vivo.10 More recent work has proposed that MCD-DLBCL instead arises from aberrant MBCs that have exited the GC reaction.11,12 Multiple recent studies have shown that introduction of the MYD88L265P or the equivalent mouse allele (Myd88L252P) into B cells in vivo results in an expansion of IgM+ PCs.13-19 When Myd88L252P was combined with conditional overexpression of BCL2 in B cells, to mimic the amplification of BCL2 that occurs in one-half of MCD cases,4 young animals exhibited a further increase in PCs.16 It is unclear whether these mutant Myd88-expressing PCs or aberrant MBCs, described in other settings, exhibit dependencies similar to MCD-derived cell lines.20
PC differentiation is thought to be initiated in the light zone (LZ) when GC B cells (GCBs) expressing higher affinity BCR experience increased BCR signaling in response to antigen.21 These cells transit to the dark zone (DZ) and receive other signals that allow them to differentiate into PBs.22,23 Increased BCR signaling promotes NF-κB activation, driving increased IRF4 expression that can then promote the expression of the PC lineage–defining transcription factor PRDM1 (Blimp-1). PRDM1 and IRF4 antagonize BCL6 and promote exit from the GC via differentiation into the PC fate.24-26PRDM1 is mutated or lost in 33% of MCD-DLBCL and can act as a B-cell tumor suppressor in vivo.27,28 In contrast to PC differentiation, the molecular cues governing MBC emergence from the GC are less clear.29 However, MBCs are thought to arise from precursor cells in the LZ and are not enriched for cells bearing high-affinity BCRs.
In this study, we sought to understand how hallmark genetic lesions found in MCD-DLBCL perturb the B-cell response in vivo to better define the premalignant cell-of-origin. We found that expression of mutant Myd88 in mature B cells resulted in a cell-intrinsic expansion in spontaneous splenic GCs but had little effect on GC expansion in other settings. As with MCD, these GCBs were enriched for BCRs with evidence of self-reactivity. Myd88-mutant IgM+ GCBs displayed increased sensitivity to ibrutinib and were dependent on Tlr9 expression. Although expression of mutant Myd88 and Cd79b promoted expansion of IgM+ splenic GCBs, it also led to accumulation of GC-derived terminally differentiated PCs. Blocking PC differentiation through loss of Prdm1 in the presence of Myd88L252P and Cd79bY195H promoted a synergistic expansion of spontaneous splenic GCBs that exhibited increased proliferation and IRF4 expression but also resulted in increased GCB death. Finally, blocking cell death through overexpression of BCL2 in the context of increased My-T-BCR signaling and a PC differentiation block resulted in a massive expansion of spontaneous splenic GCBs in young animals and the development of DLBCL in aged animals. Thus, by studying animals bearing multiple MCD-associated genetic lesions, we have identified aberrant spontaneous splenic GCBs as a likely cell-of-origin in MCD.
Methods
Animals and treatments
Myd88L252P conditional knock-in, Rosa26LSL-BCL2-IRES-GFP, S1pr2-creERT2 BAC-transgenic and Rosa26LSL-tdTomato mice have been described.17,30,31Cr2-cre, Rosa26Cas9, Prdm1f/f, and Aicdacre/cre were from The Jackson Laboratory. Cd79bY195H conditional knock-in mice were generated via homologous recombination in C57BL/6NCr embryonic stem cells. Bone marrow chimeras were made using B6-Ly5.1/Cr mice from Charles River Laboratories as hosts. Hosts were lethally irradiated with 900 rad in split doses and reconstituted by tail vein injection with at least 3 × 106 bone marrow (BM) cells from the indicated donors as previously described.32
All experiments were performed in accordance with guidelines under protocols approved by the National Cancer Institute Animal Care and Use Committee.
Flow cytometry
Peripheral lymph node (pLN), spleen, mesenteric lymph node, and Peyer’s patch (PP) cell suspensions were generated by mashing organs through 70-μm cell strainers and stained with antibodies directed against surface antigens and/or were fixed, permeabilized, and stained intracellularly. Flow cytometry was performed on a CytoFLEX LX. Cell sorting was performed on a Sony MA900.
Immunohistochemistry
Spleens or lymph nodes were fixed in paraformaldehyde, dehydrated in sucrose, cryosectioned, and stained with the indicated antibodies. Images were captured with a Keyence BZ-X800 microscope.
RNA isolation, heavy chain sequencing
GCBs or MBCs from the indicated animals were sorted directly into lysis buffer for RNA extraction. Heavy chain V usage from RNA was assessed by repertoire sequencing by iRepertoire, Inc.
TMD8 knockout rescue assay
Knockout rescue assays were performed as previously described.6
Statistical analysis
Prism software (GraphPad Software) was used for all statistical analysis. Data were analyzed with a two-sample unpaired (or paired, where indicated) t test. P values were considered significant when ≤.05.
Detailed protocols are provided in the supplemental Methods (available on the Blood Web site).
Results
Myd88L252P promotes expansion of spontaneous splenic GCBs that show evidence of oncogenic signaling in vivo
To generate animals expressing Myd88L252P in mature B cells, we crossed mice with a conditional knock-in allele of Myd88L252P to Cr2-cre mice, in which cre recombinase is expressed in mature B cells.17,33 Because most mature B-cell malignancies are thought to arise from GCBs or their progeny, we evaluated the effect of Myd88L252P on GC homeostasis in mixed BM chimeras. Because spontaneous splenic GCs are highly dependent on B-cell intrinsic Myd88, we evaluated the effect of Myd88L252P in spontaneous splenic GCs in addition to GCs in immunized pLNs and gut-associated lymphoid tissue (GALT) (Figure 1A). We found that Myd88L252P promoted outgrowths of GCs most strongly in spontaneous splenic GCs and to a lesser extent in immunized GCs in the pLN and had no effect on GCs in GALT (Figure 1B-C). In unimmunized spleens, the competitive advantage conferred by Myd88L252P was strongest in GCBs and weaker in PCs (Figure 1D). No advantage was seen among Myd88L252P-expressing marginal zone B cells.
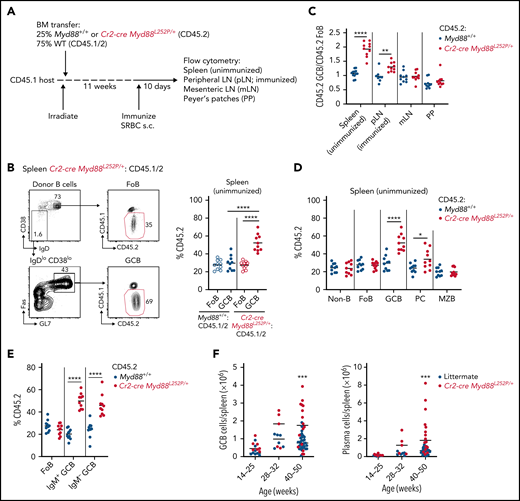
Myd88L252P promotes cell-intrinsic expansion of spontaneous splenic GCBs. (A) Experimental scheme for data in panels B to E. (B) Percentages of CD45.2 follicular B cells (FoB) and GCBs in unimmunized spleen of mixed BM chimeras generated as in panel A. Example gating strategy for FoB and GCBs is shown on left in panel B. (C) Ratio of frequency of CD45.2 GCBs to CD45.2 FoB in control or Cr2-creMyd88L252P/+ mixed BM chimeras in unimmunized spleen, SRBC-immunized pLNs, mesenteric lymph nodes (mLNs), and PPs. (D) Percentages of CD45.2 non-B cells (non-B), FoBs, GCBs, PCs, and marginal zone B cells (MZB) from mixed chimeras generated as in panel A. (E) Percentages of CD45.2 FoBs and IgM+ or IgM– GCBs in unimmunized spleen of mixed BM chimeras generated as in panel A. (F) GCBs or PCs per spleen of littermate or Cr2-cre Myd88L252P mice aged up to 1 year. Data in panels A to E are pooled from 2 independent experiments representative of 4 with 5 mice per group total. Data in panel F are from a total of 43 control and 28 Cr2-cre Myd88L252P animals. *P < .05, **P < .01, ***P < .001, **** P < .0001, unpaired two-tailed t test.
Myd88L252P promotes cell-intrinsic expansion of spontaneous splenic GCBs. (A) Experimental scheme for data in panels B to E. (B) Percentages of CD45.2 follicular B cells (FoB) and GCBs in unimmunized spleen of mixed BM chimeras generated as in panel A. Example gating strategy for FoB and GCBs is shown on left in panel B. (C) Ratio of frequency of CD45.2 GCBs to CD45.2 FoB in control or Cr2-creMyd88L252P/+ mixed BM chimeras in unimmunized spleen, SRBC-immunized pLNs, mesenteric lymph nodes (mLNs), and PPs. (D) Percentages of CD45.2 non-B cells (non-B), FoBs, GCBs, PCs, and marginal zone B cells (MZB) from mixed chimeras generated as in panel A. (E) Percentages of CD45.2 FoBs and IgM+ or IgM– GCBs in unimmunized spleen of mixed BM chimeras generated as in panel A. (F) GCBs or PCs per spleen of littermate or Cr2-cre Myd88L252P mice aged up to 1 year. Data in panels A to E are pooled from 2 independent experiments representative of 4 with 5 mice per group total. Data in panel F are from a total of 43 control and 28 Cr2-cre Myd88L252P animals. *P < .05, **P < .01, ***P < .001, **** P < .0001, unpaired two-tailed t test.
Because ABC/MCD-DLBCL is highly enriched for IgM BCRs,4,6 we assessed whether Myd88L252P was able to promote greater expansion of IgM-expressing (IgM+) GCBs compared with switched (IgM–) GCBs in unimmunized spleens of mixed chimeras. We found that there was a trend toward increased representation of Myd88L252P-expressing cells among IgM+ GCBs compared with IgM– that was not statistically significant (Figure 1E). We also aged a cohort of Cr2-cre Myd88L252P and littermate control animals for up to 1 year and found that Myd88L252P supported GC and PC expansions in the spleen (Figure 1F). These data show that Myd88L252P promotes cell-intrinsic expansions in spontaneous splenic GCs.
MCD-DLBCLs are strongly enriched for self-reactive IgM BCRs.34 Self-reactivity can occur due to interaction with self-glycoproteins, self-antigen from apoptotic cells, or binding to the BCR itself.6 Survival of MCD-DLBCL cell lines in vitro is dependent on this self-reactivity. Following depletion of endogenous heavy chain, cell survival can be rescued with expression of autologous or heterologous heavy chains from MCD-DLBCL but not with heavy chains isolated from GC-derived DLBCL. Given that outgrowths of Myd88L252P cells were most prominent in spontaneous splenic GCs compared with immunized GCs, we assessed whether heavy chain variable regions (IgVH) cloned from spontaneous splenic or immunized Myd88L252P GCB fused to the mouse IgM constant region could rescue survival of the MCD-DLBCL cell line TMD8 after CRISPR-mediated depletion of endogenous heavy chain. We found that IgVH from spontaneous splenic GCs could rescue survival of TMD8 cells, whereas IgVH from immunized GCs could not (Figure 2A-B). These data suggest that Myd88L252P preferentially promotes outgrowths in spontaneous splenic GCs due to recruitment of BCRs with similar properties to self-reactive BCRs found in MCD-DLBCL.

Myd88L252P induces MCD lymphoma-associated dependencies in spontaneous splenic GCBs. (A-B) Cas9-expressing TMD8 (MCD-DLBCL) cells were transduced with vectors expressing sgRNA directed against endogenous heavy chain and rescued with vectors expressing autologous or heterologous IgVH from BCRs from TMD8 itself or GCB-DLBCL cell lines (OCI-Ly-19 or BJAB) or spontaneous splenic or immunized GCBs from Cr2-cre Myd88L252P fused to the mouse IgM constant region; percent rescue relative to autologous heavy chain was assessed 6 days later. Data in panel B are pooled from 5 independent experiments; each bar represents a distinct heavy chain, 15 IgVH were cloned from unimmunized and 6 IgVH were cloned from SRBC-immunized Cr2-cre Myd88L252P/+ GCBs (see Methods). (C-D) Frequency of CD45.2+ cells among follicular B cells (FoB), IgM+, or IgM– GCBs in Cr2-cre Myd88L252P/+ mixed BM chimeras were treated with ibrutinib for 3 days. Data in panel D are pooled from 2 independent experiments with a total of 10 mice per group. (E-F) Irradiated hosts were reconstituted with Rosa26Cas9 or Cr2-cre Myd88L252P/+Rosa26Cas9 BM that was transduced with vectors expressing control sgRNA or sgRNA targeting Tlr9 and the fluorescent reporter Ametrine. The ratio of sgRNA+ (Ametrine+) GCBs (GCB) relative to sgRNA+ (Ametrine+) FoB in unimmunized spleen was assessed 12 weeks later. Data in panel F are pooled from 2 independent experiments with a total of 10 mice per group. (G-H) Frequency of CD45.2+ cells among FoB, IgM– GCBs, or IgM+ GCBs in Cr2-cre Myd88L252P/+ mixed BM chimeras were treated with antibiotics (ampicillin 1 g/L, vancomycin 0.5 g/L, neomycin 1 g/L, and metronidazole 1 g/L) in drinking water for 12 weeks. Data in panel H are from one experiment with 9 and 10 mice per group, respectively. *P < .05, **P < .01, ***P < .001, unpaired two-tailed t test.
Myd88L252P induces MCD lymphoma-associated dependencies in spontaneous splenic GCBs. (A-B) Cas9-expressing TMD8 (MCD-DLBCL) cells were transduced with vectors expressing sgRNA directed against endogenous heavy chain and rescued with vectors expressing autologous or heterologous IgVH from BCRs from TMD8 itself or GCB-DLBCL cell lines (OCI-Ly-19 or BJAB) or spontaneous splenic or immunized GCBs from Cr2-cre Myd88L252P fused to the mouse IgM constant region; percent rescue relative to autologous heavy chain was assessed 6 days later. Data in panel B are pooled from 5 independent experiments; each bar represents a distinct heavy chain, 15 IgVH were cloned from unimmunized and 6 IgVH were cloned from SRBC-immunized Cr2-cre Myd88L252P/+ GCBs (see Methods). (C-D) Frequency of CD45.2+ cells among follicular B cells (FoB), IgM+, or IgM– GCBs in Cr2-cre Myd88L252P/+ mixed BM chimeras were treated with ibrutinib for 3 days. Data in panel D are pooled from 2 independent experiments with a total of 10 mice per group. (E-F) Irradiated hosts were reconstituted with Rosa26Cas9 or Cr2-cre Myd88L252P/+Rosa26Cas9 BM that was transduced with vectors expressing control sgRNA or sgRNA targeting Tlr9 and the fluorescent reporter Ametrine. The ratio of sgRNA+ (Ametrine+) GCBs (GCB) relative to sgRNA+ (Ametrine+) FoB in unimmunized spleen was assessed 12 weeks later. Data in panel F are pooled from 2 independent experiments with a total of 10 mice per group. (G-H) Frequency of CD45.2+ cells among FoB, IgM– GCBs, or IgM+ GCBs in Cr2-cre Myd88L252P/+ mixed BM chimeras were treated with antibiotics (ampicillin 1 g/L, vancomycin 0.5 g/L, neomycin 1 g/L, and metronidazole 1 g/L) in drinking water for 12 weeks. Data in panel H are from one experiment with 9 and 10 mice per group, respectively. *P < .05, **P < .01, ***P < .001, unpaired two-tailed t test.
We next examined whether there was functional evidence for My-T-BCR signaling occurring in Myd88L252P-expressing B cells in spontaneous splenic GCs in vivo. MCD-DLBCL lines containing the My-T-BCR complex are highly sensitive to the Bruton’s tyrosine kinase inhibitor ibrutinib in vitro.5 To test whether spontaneous splenic GCs expressing Myd88L252P showed enhanced sensitivity to ibrutinib, we treated mixed chimeras with ibrutinib for 3 days and assessed for the frequency of Myd88L252P-expressing B cells among IgM+ or IgM– GCBs (Figure 2C). We found that the competitive advantage of Myd88L252P-expressing IgM+ GCBs was reduced in ibrutinib-treated animals (Figure 2D). TLR9, a component of the My-T-BCR complex, was required for survival of MCD-DLBCL cell lines. In contrast, Tlr9 was not required for survival of wild-type (WT) spontaneous splenic GCs and instead suppressed these GCs.9 To assess the role of Tlr9 in Myd88L252P-expressing spontaneous splenic GCs, we reconstituted irradiated hosts with BM from WT or Cr2-cre Myd88L252P mice that also constitutively expressed Cas9 (Rosa26Cas9/+ or Cr2-cre Myd88L252P Rosa26Cas9/+) transduced with retrovirus expressing Tlr9-targeting single guide RNA (sgRNA) and the fluorescent reporter Ametrine (Figure 2E). Consistent with published data, loss of Tlr9 led to outgrowths of GCB in Rosa26Cas9 chimeras (Figure 2F). In contrast, Myd88L252P-expressing IgM+ GCBs were dependent on Tlr9 expression. Collectively, these data suggest that Myd88L252P promotes aberrant signaling in spontaneous splenic GCBs with dependencies similar to MCD-DLBCL.
Given that spontaneous splenic GCs form in unimmunized animals and are dependent on Myd88, it was possible that Myd88L252P mutations might promote exaggerated GC responses to microbiota-derived ligands. To test whether commensal microbiota could drive expansion of Myd88L252P GCBs, we treated mixed chimeras with antibiotic-containing water for 12 weeks and found that commensal depletion increased representation of Myd88L252P GCB in spontaneous splenic GCs (Figure 2G-H). These data suggest that self-antigen, not microbiota-derived ligands, is principally responsible for the competitive advantage of Myd88L252P GCBs.
Myd88L252P and Cd79bY195H cooperate to promote IgM+ PB and PC output from spontaneous splenic GCs
Mutations of CD79B frequently co-occur with MYD88 mutations in both ABC-DLBCL and MCD-DLBCL.4 The most frequent CD79B alteration is a missense mutation of the first tyrosine residue (Y196) within the ITAM motif.34 Y196 mutations lead to sustained BCR signaling by reducing BCR internalization-induced negative feedback. We generated animals that conditionally express a Cd79b allele encoding Y195H, analogous to Y196H in human (supplemental Figure 1A). Mixed BM chimeras were generated from Cr2-cre Cd79bY195H animals, and we found little effect of Cd79bY195H expression alone on GC homeostasis (supplemental Figure 1B-C). We then intercrossed animals containing the Myd88L252P and Cd79bY195H conditional knock-in alleles and Cr2-cre to generate mice that express both Myd88L252P and Cd79bY195H in mature B cells (hereafter referred to as Myd88/Cd79b). We generated mixed BM chimeras and found that Myd88/Cd79b IgM+ GCBs showed stronger outgrowths compared with IgM– GCBs in unimmunized spleens (Figure 3A; supplemental Figure 1D). A small population of cells in the GC gate were noted that were positive for IRF4 and CD138, stained strongly for intracellular immunoglobulin, and displayed lower levels of B220. Because these cells expressed both GC markers and PC markers, they were likely GC-derived PBs. The competitive advantage conferred by Myd88/Cd79b in mixed chimeras was strongest in IgM+ PBs. Myd88/Cd79b IgM+ GCBs exhibited a modest increase in proliferation compared with internal controls, whereas Myd88/Cd79b IgM– GCBs did not (Figure 3B). In 6- to 9-month-old Myd88/Cd79b mice, we saw an accumulation of IgM+ PBs and PCs (Figure 3C; supplemental Figure 1E). IgM+ PBs in aged Myd88/Cd79b animals showed decreased proliferation compared with IgM+ GCBs (Figure 3D).
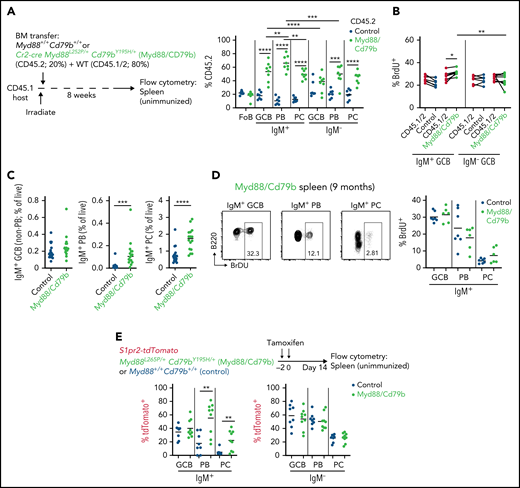
Myd88L252P and Cd79bY195H cooperate to promote IgM+ PB and PC output from spontaneous splenic GCs. (A) Percentages of CD45.2 follicular B cells (FoB), IgM– or IgM+ GCBs, PBs, or PCs in unimmunized spleens of mixed BM chimeras that were generated with a mixture of 20% Cr2-cre Myd88L252P/+Cd79bY195H/+ (Myd88/Cd79b) or control BM (CD45.2) and 80% WT CD45.1/2 eight weeks after irradiation. Example gating scheme is depicted in supplemental Figure 2A. (B) Intracellular fluorescence-activated cell sorting for 5-bromo-2′-deoxyuridine (BrdU) incorporation in IgM– or IgM+ GCBs from spleens of control or Myd88/Cd79b mixed BM chimeras that were treated intraperitoneally with BrdU 30 minutes before euthanasia. Frequency of (C) or BrdU incorporation in (D) IgM+ GCBs, IgM+ PBs, or IgM+ PCs in spleens of unimmunized control or Myd88/Cd79b mice that were 6 to 9 months old. (E) Frequency of tdTomato+ cells among IgM+ or IgM– GCs, PBs, PCs, or MBCs in S1pr2-creERT2 Rosa26LSLtdTomato/+ Myd88L252P/+ Cd79bY195H/+ or control mice 14 days after Cre induction by tamoxifen. Example gating for tdTomato is shown in supplemental Figure 2B. Data in panel A are from one experiment representative of three with 7 and 8 mice per group, respectively; data in panel B are from one experiment representative of two with 7 and 8 mice per group; data in panel C are pooled data from 8 independent experiments with 19 and 16 mice per group; data in panel D are pooled data from 4 independent experiments with 7 and 5 mice per group; data in panel E are pooled from 5 independent experiments with a total of 8 and 9 mice per group. *P < .05, **P < .01, ***P < .001, ****P < .0001, paired two-tailed t test for data in panel A comparing populations from the same mice and in panel B; and unpaired two-tailed t test for data in panel A comparing populations between mice and in panel E.
Myd88L252P and Cd79bY195H cooperate to promote IgM+ PB and PC output from spontaneous splenic GCs. (A) Percentages of CD45.2 follicular B cells (FoB), IgM– or IgM+ GCBs, PBs, or PCs in unimmunized spleens of mixed BM chimeras that were generated with a mixture of 20% Cr2-cre Myd88L252P/+Cd79bY195H/+ (Myd88/Cd79b) or control BM (CD45.2) and 80% WT CD45.1/2 eight weeks after irradiation. Example gating scheme is depicted in supplemental Figure 2A. (B) Intracellular fluorescence-activated cell sorting for 5-bromo-2′-deoxyuridine (BrdU) incorporation in IgM– or IgM+ GCBs from spleens of control or Myd88/Cd79b mixed BM chimeras that were treated intraperitoneally with BrdU 30 minutes before euthanasia. Frequency of (C) or BrdU incorporation in (D) IgM+ GCBs, IgM+ PBs, or IgM+ PCs in spleens of unimmunized control or Myd88/Cd79b mice that were 6 to 9 months old. (E) Frequency of tdTomato+ cells among IgM+ or IgM– GCs, PBs, PCs, or MBCs in S1pr2-creERT2 Rosa26LSLtdTomato/+ Myd88L252P/+ Cd79bY195H/+ or control mice 14 days after Cre induction by tamoxifen. Example gating for tdTomato is shown in supplemental Figure 2B. Data in panel A are from one experiment representative of three with 7 and 8 mice per group, respectively; data in panel B are from one experiment representative of two with 7 and 8 mice per group; data in panel C are pooled data from 8 independent experiments with 19 and 16 mice per group; data in panel D are pooled data from 4 independent experiments with 7 and 5 mice per group; data in panel E are pooled from 5 independent experiments with a total of 8 and 9 mice per group. *P < .05, **P < .01, ***P < .001, ****P < .0001, paired two-tailed t test for data in panel A comparing populations from the same mice and in panel B; and unpaired two-tailed t test for data in panel A comparing populations between mice and in panel E.
To assess whether Myd88/Cd79b conferred a greater propensity of IgM+ GCBs to differentiate into PBs and PCs, we crossed Myd88/Cd79b mice to animals carrying a GC-specific tamoxifen-inducible fate reporter allele (S1pr2-tdTomato; S1pr2-creETR2 Rosa26LSLtdTomato/+).31 In this system, after administration of tamoxifen, GCBs and their progeny express Myd88L252P, Cd79bY195H, and tdTomato. We found that Myd88/Cd79b increased GC output of IgM+ PBs and IgM+ PCs but had no effect on switched PBs or PCs (Figure 3E; supplemental Figure 1F). Collectively, these data show that although Myd88/Cd79b can modestly promote the expansion and proliferation of IgM+ GCBs, it also increases output from the GC to IgM+ PBs and PCs with decreased proliferation compared with GCBs.
Loss of Prdm1 synergizes with Myd88L252P and Cd79bY195H to promote increased proliferation, IRF4 expression, and cell death in spontaneous splenic GCs
Given that expression of Myd88L252P and Cd79bY195H promotes PC differentiation from spontaneous splenic GCs in vivo, we hypothesized that loss of Prdm1, as occurs in approximately one-third of MCD-DLBCL, might synergize with Myd88/Cd79b and promote expansion of GCBs in unimmunized splenic GCs. In mixed chimeras, loss of Prdm1 in B cells alone (Cr2-cre Prdm1f/f) resulted in a 1.6-fold GC outgrowth that was not restricted to spontaneous splenic GCs (supplemental Figure 2A-B). Prdm1 deficiency supported stronger expansions of switched GCBs compared with IgM+ GCBs in unimmunized spleens (supplemental Figure 2C). We then crossed Prdm1f/f animals to Cr2-cre and Myd88/Cd79b animals to generate Cr2-cre Myd88L252P/+ Cd79bY195H/+ Prdm1f/f animals (Myd88/Cd79b/Prdm1). We generated mixed chimeras with ∼5% Myd88/Cd79b/Prdm1 BM that was CD45.2 and ∼95% WT CD45.1/2 BM. We found that Myd88/Cd79b/Prdm1 induced a 10-fold competitive advantage in spontaneous splenic GCs (Figure 4A-C). As expected, loss of Prdm1 in the presence of Myd88/Cd79b blocked PC differentiation (Figure 4B). Myd88/Cd79b/Prdm1 cells were strongly enriched in the GC DZ and showed less representation in the LZ (Figure 4D; supplemental Figure 2D). In addition, a DZ bias was seen in unimmunized spleens of Cr2-cre Myd88L252P/+ and Myd88/Cd79b mixed chimeras (supplemental Figure 2E). Despite the lack of PC output from the GC, Myd88/Cd79b/Prdm1 did not result in a proportional increase in memory phenotype cells or in the small population of pre-memory GCBs in the LZ compared with GCBs themselves. Splenic Myd88/Cd79b/Prdm1 IgM+ GCBs displayed increased proliferation compared with both WT IgM+ GCBs and IgM– Myd88/Cd79b/Prdm1 GCBs (Figure 4E). Splenic Myd88/Cd79b/Prdm1 IgM+ GCBs exhibited increased IRF4 expression, an important readout of BCR-driven NF-κB signaling, relative to both WT internal controls and IgM– Myd88/Cd79b/Prdm1 cells (Figure 4F). Importantly, in immunized pLNs, there was only a modest increase in proliferation of mutant GCBs compared with WT cells, with no difference between IgM+ and IgM– Myd88/Cd79b/Prdm1 GCBs, and there was no increase in IRF4 in IgM+ Myd88/Cd79b/Prdm1 GCBs (supplemental Figure 2F-G). These data are consistent with the hypothesis that IgM+ BCRs with intrinsic signaling properties such as self-reactivity are preferentially recruited to spontaneous splenic GCs to support GC expansion at these sites in the presence of MCD-associated genetic alterations.

Loss of Prdm1 synergizes with Myd88L252P and Cd79bY195H to promote outgrowths in spontaneous splenic GCs. (A) Experimental scheme for data in panels B to H. Percentages of CD45.2 follicular B cells (FoB), GCBs, or PCs in unimmunized spleens (B) or ratios of frequency of CD45.2 GCB to CD45.2 follicular B cells (C) in unimmunized spleens, immunized pLNs, mesenteric lymph nodes (mLNs), or PPs of mixed BM chimeras that were generated with a mixture of ∼5% Cr2-cre Myd88L252P/+Cd79bY195H/+Prdm1f/f (Myd88/Cd79b/Prdm1) or control BM (CD45.2) and ∼95% WT CD45.1/2 eight weeks after irradiation. (D) Frequency of CD45.2 cells among FoB, DZ GCBs, LZ GCBs, pre-memory GCBs, or MBCs in Myd88/Cd79b/Prdm1 mixed chimeras. Example gating strategy is shown in supplemental Figure 3D. Intracellular fluorescence-activated cell sorting for 5-bromo-2′-deoxyuridine (BrdU) incorporation (E) or IRF4 expression (F) in IgM+ or IgM– GCBs from spleens of control or Myd88/Cd79b/Prdm1 mixed BM chimeras that were treated intraperitoneally with BrdU 30 minutes before euthanasia. (G) Frequency of CD45.2 IgM+ or IgM– GCBs in spleens of control or Myd88/Cd79b/Prdm1 mixed chimeras. (H) Percentage of active-Caspase-3+ GCBs in control, Myd88/Cd79b, or Myd88/Cd79b/Prdm1 mixed chimeras. Example gating strategy is shown on the left. Data in panels B, C, and E to H are pooled from 3 to 5 independent experiments with a total of at least 11 mice per group. Data in panel D are pooled from 2 experiments with 6 and 9 mice per group, respectively. *P < .05, **P < .01, ****P < .0001 paired two-tailed t test for data in panels E, F, and H. *P < .05, **P<.01, ***P < .001, ****P < .0001, unpaired two-tailed t test for all other data. MFI, mean fluorescence intensity.
Loss of Prdm1 synergizes with Myd88L252P and Cd79bY195H to promote outgrowths in spontaneous splenic GCs. (A) Experimental scheme for data in panels B to H. Percentages of CD45.2 follicular B cells (FoB), GCBs, or PCs in unimmunized spleens (B) or ratios of frequency of CD45.2 GCB to CD45.2 follicular B cells (C) in unimmunized spleens, immunized pLNs, mesenteric lymph nodes (mLNs), or PPs of mixed BM chimeras that were generated with a mixture of ∼5% Cr2-cre Myd88L252P/+Cd79bY195H/+Prdm1f/f (Myd88/Cd79b/Prdm1) or control BM (CD45.2) and ∼95% WT CD45.1/2 eight weeks after irradiation. (D) Frequency of CD45.2 cells among FoB, DZ GCBs, LZ GCBs, pre-memory GCBs, or MBCs in Myd88/Cd79b/Prdm1 mixed chimeras. Example gating strategy is shown in supplemental Figure 3D. Intracellular fluorescence-activated cell sorting for 5-bromo-2′-deoxyuridine (BrdU) incorporation (E) or IRF4 expression (F) in IgM+ or IgM– GCBs from spleens of control or Myd88/Cd79b/Prdm1 mixed BM chimeras that were treated intraperitoneally with BrdU 30 minutes before euthanasia. (G) Frequency of CD45.2 IgM+ or IgM– GCBs in spleens of control or Myd88/Cd79b/Prdm1 mixed chimeras. (H) Percentage of active-Caspase-3+ GCBs in control, Myd88/Cd79b, or Myd88/Cd79b/Prdm1 mixed chimeras. Example gating strategy is shown on the left. Data in panels B, C, and E to H are pooled from 3 to 5 independent experiments with a total of at least 11 mice per group. Data in panel D are pooled from 2 experiments with 6 and 9 mice per group, respectively. *P < .05, **P < .01, ****P < .0001 paired two-tailed t test for data in panels E, F, and H. *P < .05, **P<.01, ***P < .001, ****P < .0001, unpaired two-tailed t test for all other data. MFI, mean fluorescence intensity.
Despite the increase in proliferation and IRF4 expression among IgM+ Myd88/Cd79b/Prdm1 GCBs, IgM+ GCBs were not expanded compared with IgM– GCBs in Myd88/Cd79b/Prdm1 mixed chimeras (Figure 4G). In addition, we did not observe gross histologic alterations in Myd88/Cd79b/Prdm1 animals that were aged to 6 months. We hypothesized that this lack of expansion of IgM+ GC cells and the lack of gross histologic changes could be due to increased cell death among Myd88/Cd79b/Prdm1 GCBs. It is estimated that approximately one-half of GCBs undergo cell death every 6 hours to maintain GC size given the high proliferative rate of GCBs.35 Only a small fraction of dying cells can be detected ex vivo by fluorescence-activated cell sorting due to rapid uptake of apoptotic GCBs by tingible body macrophages in vivo. We analyzed apoptosis ex vivo by fluorescence-activated cell sorting in spontaneous splenic GCs from mixed chimeras and found that Myd88/Cd79b/Prdm1 (and to a lesser extent Myd88/Cd79b alone) promoted a highly significant increase in cell death relative to WT GCBs (Figure 4H).
BCL2 overexpression synergizes with Myd88L252P and Cd79bY195H and loss of Prdm1 to promote massive expansion of spontaneous splenic GCs and DLBCL
Increased GCB death as a result of Myd88/Cd79b/Prdm1 may occur as a compensatory response to increased My-T-BCR–driven signaling when PC differentiation is blocked. Amplifications of the antiapoptotic molecule BCL2 occur in one-half of MCD-DLBCL cases.4 Our data suggest that BCL2 amplification may be selected for in MCD to protect from My-T-BCR–driven cell death. Therefore, we crossed animals carrying an allele that conditionally overexpresses BCL2 and a green fluorescent protein (GFP) reporter (Rosa26LSL-BCL2-IRES-GFP) to animals carrying Myd88/Cd79b/Prdm1 and Aicdacre.17,36 We used Aicdacre to limit BCL2 overexpression to pre-GCBs and their progeny. Unimmunized spleens from Myd88/Cd79b/Prdm1/BCL2 or littermate control animals at 8 weeks of age by were analyzed by immunohistochemistry. Staining with GL7 showed that Myd88/Cd79b/Prdm1/Bcl2 animals had a dramatic increase in GC size and number compared with all other genotypes. GCs from Myd88/Cd79b/Prdm1/BCL2 were bizarrely shaped and some had minimal mantle areas, appearing to extend into the red pulp (Figure 5A, top row; supplemental Figure 3A). CD138 staining showed increased PCs in Myd88/Cd79b/BCL2 animals that were completely lost in Myd88/Cd79b/Prdm1/BCL2 animals (Figure 5A, second row). GCs in Myd88/Cd79b/Prdm1/BCL2 and Myd88/Cd79b/BCL2 showed decreased GL7 staining toward the center of the GC structure; however, staining with the GC marker Ephrin-B1 confirmed that cells with decreased GL7 intensity retained a GC phenotype (Figure 5A, third row; supplemental Figure 4). GL7 staining was largely coincident with CD35+ follicular dendritic cell (FDC) meshworks (Figure 5A-B). However, rare GL7+ cells could be seen extending into T-cell areas where FDC meshworks are absent (Figure 5B). Myd88/Cd79b/Prdm1/BCL2 animals displayed enlarged spleens (supplemental Figure 3B-C). Flow cytometry revealed that, although reporter-positive cells in Myd88/Cd79b/Prdm1/BCL2 animals did not downregulate CD38, approximately two-thirds of cells were Fashi and GL7+, consistent with a GC phenotype, whereas the remaining Fasint GL7– cells appeared to have an MBC phenotype (Figure 5C). GC phenotype cells from Myd88/Cd79b/Prdm1/BCL2 spleens were Ephrin-B1+ and CXCR4hi, whereas memory phenotype cells were enriched for the memory marker CCR6 (Figure 5D). Myd88/Cd79b/Prdm1/BCL2 GCBs displayed a strong bias toward the CXCR4hiCD86lo DZ immunophenotype (Figure 5E). GCBs were twice as abundant as MBCs in Myd88/Cd79b/Prdm1/BCL2 animals and were expanded compared with other genotypes (Figure 5F; supplemental Figure 3D). To test whether this constellation of genetic changes could drive tumorigenesis in vivo, we analyzed Myd88/Cd79b/BCL2 or Myd88/Cd79b/Prdm1/BCL2 full BM chimeras 6 to 8 months after reconstitution. In all Myd88/Cd79b/BCL2 animals, there was expansion of GCs and PCs histologically (Figure 6A; supplemental Figure 4). GCs were coincident with FDC meshworks, and there was maintenance of lymphoid architecture consistent with lymphoid hyperplasia (Figure 6A-B; supplemental Figure 4). In contrast, 5 of 8 Myd88/Cd79b/Prdm1/BCL2 animals showed areas of effacement of splenic architecture with expansion of the white pulp due to a proliferation of large atypical lymphoid cells (Figure 6A; supplemental Figure 5). Areas with effacement stained positive for GL7, B220, and Ki-67; were devoid of FDC meshworks; and were negative for CD138, consistent with DLBCL. Two of 5 animals with splenic tumors also displayed retroperitoneal tumors involving lymph node or accessory splenic tissue (supplemental Figure 5). Myd88/Cd79b/Prdm1/BCL2 animals that lacked tumors showed partial loss of GCs but had varying degrees of GL7+ B cells in T-cell areas similar to the infiltration of GL7+ cells in young animals (compare nontumor-bearing mice in supplemental Figure 5 vs Figure 5B). In tumor-bearing mice, residual follicles containing naive IgD+ B cells and remnants of FDC meshworks could be seen surrounding DLBCL areas, suggesting that tumors arose from GCBs that had previously infiltrated T-cell areas.

Overexpression of BCL2 cooperates with Myd88/Cd79b/Prdm1 to induce massive expansion of aberrant spontaneous splenic GCBs. (A) Immunohistochemistry of unimmunized spleens from 8-week-old mice that were Aicdacre/+ and control, Rosa26LSLBCL2-IRES-GFP (BCL2), Myd88/Cd79b/Prdm1, Myd88/Cd79b/BCL2, or Myd88/Cd79b/Prm1/BCL2. Sections were stained for IgD and GL7, CD138, Ephrin-B1, or CD35. Scale bar = 500 μm. Additional examples of Aicdacre/+ Myd88/Cd79b/BCL2 or Aicdacre/+ Myd88/Cd79b/Prdm1/BCL2 mice are shown in supplemental Figure 3A. (B) High-power images of areas marked in panel A. Sections were stained for IgD and GL7, CD35, or CD4. Red asterisks mark areas of GL7+ cell infiltration into T-cell areas of spleens of Myd88/Cd79b/Prdm1/BCL2 animals. Scale bar = 100 μm. (C) Flow cytometry of splenocytes from Aicdacre/+ Myd88/Cd79b/BCL2 or Aicdacre/+ Myd88/Cd79b/Prdm1/BCL2 mice. (D) Expression of Ephrin-B1, CXCR4, or CCR6 on splenic GCs or MBCs from Aicdacre/+ Myd88/Cd79b/Prdm1/BCL2 mice. (E) Expression of LZ or DZ GC markers on splenic GCBs from Aicdacre/+ control or Myd88/Cd79b/Prdm1/BCL2 mice. (F) Frequency of splenic GCBs, MBCs, or PBs/PCs in 8- to 10-week-old Aicdacre/+ control, Myd88/Cd79b/Prdm1, Myd88/Cd79b/BCL2, or Myd88/Cd79b/Prdm1/BCL2. Data in panels A and B are representative of 2 to 5 mice per genotype; data in panels C to E are from one experiment representative of 4 independent experiments with 1 mouse per group; and data in panel F are pooled from 8 independent experiments with up to 1 mouse per group per experiment. **P < .01, ***P < .001, ****P < .0001, unpaired two-tailed t test.
Overexpression of BCL2 cooperates with Myd88/Cd79b/Prdm1 to induce massive expansion of aberrant spontaneous splenic GCBs. (A) Immunohistochemistry of unimmunized spleens from 8-week-old mice that were Aicdacre/+ and control, Rosa26LSLBCL2-IRES-GFP (BCL2), Myd88/Cd79b/Prdm1, Myd88/Cd79b/BCL2, or Myd88/Cd79b/Prm1/BCL2. Sections were stained for IgD and GL7, CD138, Ephrin-B1, or CD35. Scale bar = 500 μm. Additional examples of Aicdacre/+ Myd88/Cd79b/BCL2 or Aicdacre/+ Myd88/Cd79b/Prdm1/BCL2 mice are shown in supplemental Figure 3A. (B) High-power images of areas marked in panel A. Sections were stained for IgD and GL7, CD35, or CD4. Red asterisks mark areas of GL7+ cell infiltration into T-cell areas of spleens of Myd88/Cd79b/Prdm1/BCL2 animals. Scale bar = 100 μm. (C) Flow cytometry of splenocytes from Aicdacre/+ Myd88/Cd79b/BCL2 or Aicdacre/+ Myd88/Cd79b/Prdm1/BCL2 mice. (D) Expression of Ephrin-B1, CXCR4, or CCR6 on splenic GCs or MBCs from Aicdacre/+ Myd88/Cd79b/Prdm1/BCL2 mice. (E) Expression of LZ or DZ GC markers on splenic GCBs from Aicdacre/+ control or Myd88/Cd79b/Prdm1/BCL2 mice. (F) Frequency of splenic GCBs, MBCs, or PBs/PCs in 8- to 10-week-old Aicdacre/+ control, Myd88/Cd79b/Prdm1, Myd88/Cd79b/BCL2, or Myd88/Cd79b/Prdm1/BCL2. Data in panels A and B are representative of 2 to 5 mice per genotype; data in panels C to E are from one experiment representative of 4 independent experiments with 1 mouse per group; and data in panel F are pooled from 8 independent experiments with up to 1 mouse per group per experiment. **P < .01, ***P < .001, ****P < .0001, unpaired two-tailed t test.
![MCD-associated genetic alterations promote DLBCL in vivo. (A) Spleen image and histologic analysis of 8-month-old Myd88/Cd79b/BCL2 or Myd88/Cd79b/Prdm1/BCL2 full BM chimeras. Sections were stained with hematoxylin and eosin (H&E) or for IgD and GL7, CD35, B220, CD138, or Ki-67. Scale bar is 1 cm in spleen images, 500 μM in low-power H&E and immunohistochemistry images, and 20 μm in high-power H&E (H&E [HPF]). Additional examples of Myd88/Cd79b/BCL2 or Myd88/Cd79b/Prdm1/BCL2 animals are shown in supplemental Figures 4 and 5, respectively. (B) Frequency of lymphoid hyperplasia or DLBCL in 6- to 8-month-old Myd88/Cd79b/BCL2 or Myd88/Cd79b/Prdm1/BCL2 full BM chimeras. (C) VH usage from repertoire sequencing of sorted GCBs or MBCs from tumor-bearing Myd88/Cd79b/Prdm1/BCL2 animals. (D) V-region mutation frequency per read of monoclonal outgrowths in GCBs of tumor-bearing Myd88/Cd79b/Prdm1/BCL2 animals. Dominant CDR3 peptide sequence of monoclonal outgrowth is indicated.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/140/10/10.1182_blood.2022015926/3/m_bloodbld2022015926f6a.png?Expires=1767713992&Signature=GqMZ0GD0l0y4khBn5iiWvGL8as55tP6CpOWuI68g1ndsfikPq9GUQPoT9OWYfTH06t4NJK0RH7X2p7rXuoE6NKKd6fGbLlYQeFB7wYvfHlUtqp~Z4t2tyeRhsyIAlE0EayOHuwZCWJUMYD9PF4~HxGxVchgIEXOvvi1vJ0gOyvTgEoNydPAXSD14u3krJVWeRthjRfManWvhjW9sXMZuKPhMtPM1BVkBpZopBJJ8qqtdoVBEFySzYBL40DDXsAdz3uDWcLbtx4norq84a29Liv7rPsCaqvqSL6~xGc7h4xBtYBT0ZoYLYsAXbgxkyFMAad2MvxtLHYif1oP9lf5GwA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![MCD-associated genetic alterations promote DLBCL in vivo. (A) Spleen image and histologic analysis of 8-month-old Myd88/Cd79b/BCL2 or Myd88/Cd79b/Prdm1/BCL2 full BM chimeras. Sections were stained with hematoxylin and eosin (H&E) or for IgD and GL7, CD35, B220, CD138, or Ki-67. Scale bar is 1 cm in spleen images, 500 μM in low-power H&E and immunohistochemistry images, and 20 μm in high-power H&E (H&E [HPF]). Additional examples of Myd88/Cd79b/BCL2 or Myd88/Cd79b/Prdm1/BCL2 animals are shown in supplemental Figures 4 and 5, respectively. (B) Frequency of lymphoid hyperplasia or DLBCL in 6- to 8-month-old Myd88/Cd79b/BCL2 or Myd88/Cd79b/Prdm1/BCL2 full BM chimeras. (C) VH usage from repertoire sequencing of sorted GCBs or MBCs from tumor-bearing Myd88/Cd79b/Prdm1/BCL2 animals. (D) V-region mutation frequency per read of monoclonal outgrowths in GCBs of tumor-bearing Myd88/Cd79b/Prdm1/BCL2 animals. Dominant CDR3 peptide sequence of monoclonal outgrowth is indicated.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/140/10/10.1182_blood.2022015926/3/m_bloodbld2022015926f6b.png?Expires=1767713992&Signature=tg5bBaN-N97Vr1ouWn6QTjdtdtq2j2YskUaenNGX4JOd9rBhnbSLrrdTcRMCGeHb3y-7rGYP6TUau6Fn7GZ~fb8rBb1CtHG7tD0sWTyvVZM5R-dUsqa6zSO5-PrT8QV9lfysfJ51ZlbFDSVyIUKChsP2E7iiwTNt949bkdNXIQrLQTIUatdFXcndNY47CoBIw7XJZ-cZxs0ozBifm4Ntf2amRggC9i-CyMWO2VFPpRZAu00-Mkivp8Tf5M~xu5vWedWrxb8~Wrg0RABbYUQB96Xmex91hZmRK6ZI3RNk-pxeWVl1JqMPEnFhZvNoADZ8jXuu3u2UVTD4SwopJrTKsA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
MCD-associated genetic alterations promote DLBCL in vivo. (A) Spleen image and histologic analysis of 8-month-old Myd88/Cd79b/BCL2 or Myd88/Cd79b/Prdm1/BCL2 full BM chimeras. Sections were stained with hematoxylin and eosin (H&E) or for IgD and GL7, CD35, B220, CD138, or Ki-67. Scale bar is 1 cm in spleen images, 500 μM in low-power H&E and immunohistochemistry images, and 20 μm in high-power H&E (H&E [HPF]). Additional examples of Myd88/Cd79b/BCL2 or Myd88/Cd79b/Prdm1/BCL2 animals are shown in supplemental Figures 4 and 5, respectively. (B) Frequency of lymphoid hyperplasia or DLBCL in 6- to 8-month-old Myd88/Cd79b/BCL2 or Myd88/Cd79b/Prdm1/BCL2 full BM chimeras. (C) VH usage from repertoire sequencing of sorted GCBs or MBCs from tumor-bearing Myd88/Cd79b/Prdm1/BCL2 animals. (D) V-region mutation frequency per read of monoclonal outgrowths in GCBs of tumor-bearing Myd88/Cd79b/Prdm1/BCL2 animals. Dominant CDR3 peptide sequence of monoclonal outgrowth is indicated.
MCD-associated genetic alterations promote DLBCL in vivo. (A) Spleen image and histologic analysis of 8-month-old Myd88/Cd79b/BCL2 or Myd88/Cd79b/Prdm1/BCL2 full BM chimeras. Sections were stained with hematoxylin and eosin (H&E) or for IgD and GL7, CD35, B220, CD138, or Ki-67. Scale bar is 1 cm in spleen images, 500 μM in low-power H&E and immunohistochemistry images, and 20 μm in high-power H&E (H&E [HPF]). Additional examples of Myd88/Cd79b/BCL2 or Myd88/Cd79b/Prdm1/BCL2 animals are shown in supplemental Figures 4 and 5, respectively. (B) Frequency of lymphoid hyperplasia or DLBCL in 6- to 8-month-old Myd88/Cd79b/BCL2 or Myd88/Cd79b/Prdm1/BCL2 full BM chimeras. (C) VH usage from repertoire sequencing of sorted GCBs or MBCs from tumor-bearing Myd88/Cd79b/Prdm1/BCL2 animals. (D) V-region mutation frequency per read of monoclonal outgrowths in GCBs of tumor-bearing Myd88/Cd79b/Prdm1/BCL2 animals. Dominant CDR3 peptide sequence of monoclonal outgrowth is indicated.
We performed repertoire sequencing of IgVH of GCBs and MBCs sorted from tumors. In GCBs, single VH segments comprised between 76% and 94% of reads, were monoclonal, and had undergone somatic hypermutation (Figure 6C-D). MBCs were not dominated by the monoclonal VH segments seen in GCBs (Figure 6C). Collectively, these data show that rescue of cell death by BCL2 overexpression in the context of increased My-T-BCR signaling and a blockade of PC differentiation results in a synergistic expansion of spontaneous splenic GCs in young mice and development of DLBCL upon aging.
Discussion
The current study found that MCD-associated genetic alterations perturbed homeostasis of spontaneous splenic GCBs. Expression of Myd88L252P resulted in a cell-intrinsic expansion of spontaneous splenic GCBs that displayed in vivo dependencies consistent with pathologic My-T-BCR signaling. Coexpression of Myd88L252P and Cd79bY195H resulted in increased output of IgM+ PBs and PCs from spontaneous splenic GCs. Blockade of PC differentiation through loss of Prdm1 in the presence of mutant Myd88 and Cd79b resulted in a synergistic competitive advantage in spontaneous splenic GCs. IgM+ Myd88/Cd79b/Prdm1 splenic GCBs exhibited increased proliferation and increased IRF4 but also showed increased GCB apoptosis, prompting us to generate Myd88/Cd79b/Prdm1 animals that conditionally overexpressed BCL2. Myd88/Cd79b/Prdm1/BCL2 animals showed massive expansion of splenic GCBs and later developed DLBCL.
Although GC function is most often studied in the context of immunization or infection, GCs can form in the spleen and GALT at homeostasis. GCs forming at homeostasis have unique dependencies that may be important cooperative drivers in the context of certain oncogenic events. Previous studies have highlighted the mesenteric lymph node as a site in which loss of particular tumor suppressors can more strongly drive GC expansion.37,38 The current study highlights spontaneous splenic GCs as a site that may drive the expansion of MCD-DLBCL precursor cells and support a model in which B cells expressing self-reactive or auto-reactive BCRs are preferentially recruited into these GCs compared with immunized GCs or GCs in GALT. These self-reactive BCRs cooperate with Myd88L252P to promote My-T-BCR–dependent signaling and drive both GC outgrowths and PC output from GCs. Our data suggest that MCD tumor development is restricted by the requirement for selection into the GC of premalignant B cells bearing autoreactive IgM BCRs. Further work is needed to establish the nature of self-reactivity and/or antigen-specificity of BCRs recruited to these sites and to determine whether there are specific microenvironmental cues in the unimmunized spleen that may be important for promoting or suppressing the development of malignancy at this site.39-41
It has been inferred from lymphoma genetics that MCD-DLBCL arises from B cells in the process of exiting the GC reaction but whose terminal differentiation into PCs has been stopped; however, the in vivo premalignant counterpart of these cells is unclear. Recent work has proposed that MCD-DLBCL arises from aberrant MBCs based on the accumulation of MBCs in mice with Tbl1xr1 mutations and because TBL1XR1 mutations are seen in approximately one-third of MCD-DLBCL.11,12 MBCs are derived from precursor cells present in the GC LZ.42 We find in young Myd88/Cd79b/Prdm1/BCL2 animals that there is a massive accumulation of spontaneous splenic GCBs with a DZ immunophenotype that greatly exceeds the number of MBCs, rather than decreased GCs and increased memory as occurs in Tbl1xr1-deficient settings. Moreover, gene expression studies of MCD-DLBCL do not show enrichment of MBC signatures.4 The strong accumulation of GCBs in young mice, GL7 expression on tumors of aged mice, and monoclonality of GCBs but not MBCs from tumor-bearing mice in our model support the hypothesis that aberrant GCBs and not MBCs are the likely cell-of-origin in MCD-DLBCL. Another genetic subset of ABC-DLBCL, N1 (enriched for NOTCH1 mutations), does share gene expression patterns with MBCs and is also enriched for TBL1XR1 mutations. Thus, similar to the genetic heterogeneity within ABC-DLBCL, heterogeneity likely also exists for the cell-of-origin for different genetic subtypes of ABC-DLBCL.
The development of DLBCL in approximately one-half of aged Myd88/Cd79b/Prdm1/BCL2 animals suggests that, beyond selection for a self-reactive BCR, additional genetic alterations are required to evade tumor-suppressive cues. It is known that the vast majority of MCD tumors display evidence of immune evasion through loss of expression of major histocompatibility complex class I or other proteins involved in antigen presentation.43 Although major histocompatibility complex class I deficiency alone has no effect on GC development,44 it is possible that the infiltration of GL7+ cells into T-cell areas as occurs in Myd88/Cd79b/Prdm1/BCL2 animals might enable for surveillance of premalignant cells by CD8+ T cells, leading to the depletion of GCs that occurs in aged non-tumor bearing Myd88/Cd79b/Prdm1/BCL2 animals. In future work, we plan to use Myd88/Cd79b/Prdm1/BCL2 animals to test the role of immunosurveillance in suppressing lymphoma development to establish the minimal genetic requirements for the development of malignancy in a young animal.
Acknowledgments
The authors thank L. Staudt for critical discussions; J. Cyster, J. Phelan, A. Reboldi, C. Mayer, M. Mintz, and C. Wu for providing comments on the manuscript; G. Smith for assistance with hematoxylin and eosin staining; C. Wu and Z. Chen for assistance with microscopy; and T. Ciucci and R. Bosselut for providing Prdm1f/f animals. The visual abstract was created with BioRender.com.
This research was supported by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, National Cancer Institute (1ZIABC011772-05). Funding to H.C.R. was provided by the Deutsche Forschungsgemeinschaft (RE 2246/13-1 and SFB1530-A01) and the Deutsche Krebshilfe (1117240 and 70113041). Funding to H.K. was provided by SFB1530 (Project number 455784452).
Authorship
Contribution: G.M.P, R.R., R.N.T., and H.T.N. designed and performed experiments, analyzed and interpreted data, and edited the manuscript; M.L. performed experiments and generated reagents; S.A. and V.M.M. designed and performed experiments, and analyzed and interpreted data; B.H., T.O., H.C.R., G.K., and H.K. provided key reagents; R.M.Y. designed experiments, provided key reagents, and edited the manuscript; S.P. analyzed and interpreted data and edited the manuscript; and J.R.M. designed and performed experiments, analyzed and interpreted data, and wrote the manuscript.
Conflict-of-interest disclosure: H.C.R. received consulting and lecture fees from AbbVie, AstraZeneca, Vertex, and Merck; received research funding from Gilead Pharmaceuticals; and is a cofounder of CDL Therapeutics GmbH. The remaining authors declare no competing financial interests.
Correspondence: Jagan R. Muppidi, Lymphoid Malignancies Branch, National Cancer Institute, 9000 Rockville Pike, Building 10, Room 4N/109, Bethesda, MD 20892; e-mail: jagan.muppidi@nih.gov.
All data are included in the main text or supplemental Materials.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal