

Key Points
Germline RPA1 gain-of-function missense mutations result in a telomere biology disorder phenotype.
Somatic rescue events arise in hematopoiesis secondary to germline RPA1 mutation.

Abstract
Human telomere biology disorders (TBD)/short telomere syndromes (STS) are heterogeneous disorders caused by inherited loss-of-function mutations in telomere-associated genes. Here, we identify 3 germline heterozygous missense variants in the RPA1 gene in 4 unrelated probands presenting with short telomeres and varying clinical features of TBD/STS, including bone marrow failure, myelodysplastic syndrome, T- and B-cell lymphopenia, pulmonary fibrosis, or skin manifestations. All variants cluster to DNA-binding domain A of RPA1 protein. RPA1 is a single-strand DNA-binding protein required for DNA replication and repair and involved in telomere maintenance. We showed that RPA1E240K and RPA1V227A proteins exhibit increased binding to single-strand and telomeric DNA, implying a gain in DNA-binding function, whereas RPA1T270A has binding properties similar to wild-type protein. To study the mutational effect in a cellular system, CRISPR/Cas9 was used to knock-in the RPA1E240K mutation into healthy inducible pluripotent stem cells. This resulted in severe telomere shortening and impaired hematopoietic differentiation. Furthermore, in patients with RPA1E240K, we discovered somatic genetic rescue in hematopoietic cells due to an acquired truncating cis RPA1 mutation or a uniparental isodisomy 17p with loss of mutant allele, coinciding with stabilized blood counts. Using single-cell sequencing, the 2 somatic genetic rescue events were proven to be independently acquired in hematopoietic stem cells. In summary, we describe the first human disease caused by germline RPA1 variants in individuals with TBD/STS.
Introduction
Telomeres are complex structures made up of repetitive DNA sequences associated with specialized proteins found at natural ends of linear chromosomes in all mammals. The hallmark function of telomeres is to protect chromosomal ends from degradation and inappropriate recombination, and activation of the DNA damage response.1 In the absence of telomere-associated proteins, chromosomal ends undergo premature attrition with pathologic consequence of telomere biology disorders (TBD), also referred to as short telomere syndromes (STS), dyskeratosis congenita or telomeropathies. TBD/STS are hereditary disorders that manifest on a wide phenotypic and age spectrum because of genetic heterogeneity and variable expressivity. TBD/STS–associated features include bone marrow failure (BMF), pulmonary and liver fibrosis, mucocutaneous fragility, and predisposition to myelodysplastic syndromes (MDS) and cancer.2-5 Monoallelic or biallelic inactivation in 14 genes that code for telomerase holoenzyme, shelterin complex, telomere capping machinery, and accessory telomere processes have been identified as causing TBD/STS.2,6 However, an estimated 30% of individuals with TBD/STS do not have a genetic resolve, which obscures timely diagnosis and clinical management.6
Replication protein A (RPA) is a ubiquitous, single-strand DNA (ssDNA)-binding protein that exists as a heterotrimer complex composed of RPA1, RPA2, and RPA3 in eukaryotes.7 RPA is essential for DNA replication and repair. It binds all ssDNA sequences with high affinity to provide nuclease protection, prevent hairpin formation, and recruit numerous proteins to facilitate ssDNA processing and repair. Importantly, RPA is involved in several DNA damage signaling and repair pathways, including nucleotide excision repair, base excision repair, mismatch repair, and double-strand break repair.8-11 RPA1, the largest subunit of RPA, harbors four DNA binding domains (DBD-F, DBD-A, DBD-B, and DBD-C), of which DBD-A and DBD-B are important for high-affinity ssDNA binding and facilitating protein–protein interactions.12,13 Despite no clear association with human disease, the ubiquitous involvement of RPA1 in genome integrity and the demonstrated overexpression in some cancers14-17 has driven research efforts to understand its functions. Thus far, only loss-of-function RPA1 alleles have been identified through mutagenic screens in eukaryotic systems.18-20 Disrupted DNA repair and chromosomal rearrangements were shown in yeast and human cell lines,18-20 whereas genomic instability and tumorigenesis were reported in mice.21,22 Studies in yeast revealed that RPA complex binds to telomeric regions during S phase and unfolds G-quadruplex secondary DNA structures enriched at telomeres.23-28 Furthermore, RPA1 mutants were associated with telomere shortening in yeast models and cell lines.24,25,27,29-32 However, the mechanism of how RPA1 genetic disruption affects telomere length is not well understood.
RPA1 mutations have not yet been reported in human syndromes. Here, we describe 3 germline heterozygous missense variants in RPA1 in 4 unrelated individuals with short telomeres and phenotypes seen in TBD/STS, including hematologic, pulmonary, and skin manifestations. Biochemical studies revealed that these mutants have core DNA-binding domains with elevated affinity, pointing to a unique gain-of-function (GOF) effect for at least 2 of the mutants studied, whereas cellular studies show defective hematopoiesis with telomere shortening in RPA1-mutated induced pluripotent stem cells (iPSCs). Finally, we found independently acquired somatic rescue events in 1 patient, resulting in inactivation of the germline RPA1 mutation.
Materials and methods
Patient cohort
This study was approved by the institutional review boards of the respective institutions (St Jude Children’s Hospital, INSIGHT-HD; University of Freiburg, CPMP/ICH/135/95 and 430/16; Baylor College of Medicine, H-34433; and HUPNVS, Paris 7 University, AP-HP, Institutional Review Board 00006477). Written informed consent was obtained from patients or guardians. Patient 1 (P1) and patient 2 (P2) were enrolled in the observational study of the European Working Group for MDS in Childhood (EWOG-MDS; NCT00662090). Patient 3 (P3) had an institutional diagnosis of idiopathic pulmonary fibrosis (IPF) and was enrolled at Paris 7 University, AP-HP (Institutional Review Board protocol 00006477). Patient 4 (P4) was enrolled in the Undiagnosed Diseases Network (National Institutes of Health Institutional Review Board protocol 15-HG-0130). Peripheral blood, bone marrow (BM), fibroblasts, and hair follicles were collected from patients and family members when available.
Whole-exome sequencing
Whole-exome sequencing (WES) was performed on the genomic DNA of BM and peripheral blood cells of P1 to P4 and family members (P1, parents and unaffected brother; P2, parents and unaffected sister; and P4, parents). Standard WES sequencing and analytic approaches were applied. In detail, TruSeq DNA Exome Kit (20020615; Illumina), SureSelect V5 (5190-6209; Agilent), and Human Comprehensive Exome (TWIST Biosciences) were used for enrichment and library preparation according to the manufacturers’ instructions. The generated libraries were sequenced on the Illumina HiSeq 2500 with 150 bp paired-end reads and average 50× to 100× coverage. After demultiplexing, the FASTQ files were trimmed for adapter sequences and mapped either to GRCh38/hg38 (P1_quartet) or GRCh37/hg19 (P2_quartet, P3, P4_trio) using BWA MEM (version 0.7.15). The mapping data from the P1_trio, P2_quartet, and P3 were postprocessed according to GATK best practices (GATK version 4.1.7; SAMtools and PICARD version 2.23.0), and variant calling (single nucleotide variants and short insertions/deletions) was performed by applying GATK HaplotypeCaller version 4 for the P1_trio and P3 and DeepVariant (version 1.1.0; ES mode) for the P2_quartet. The called variants were then annotated by using ANNOVAR for P1_trio and variant effector predictor (version 100) for the P2_quartet. For P3, the variant annotation and filtering processes were performed by using the Polyweb software interface of the Bioinformatics Department of Imagine Institute (Paris, France). Finally, for the P4_trio, the mapping, processing, variant calling, annotation, and filtering were achieved by using the Baylor College of Medicine institutional pipeline.33 After mapping, variant calling, and annotation, we used a single stringent filtering approach to establish a list of rare candidate variants fulfilling the following criteria: (1) minor allelic frequency below 0.01% in the Genome Aggregation Database (gnomAD) population; (2) minimum 6 altered forward and reverse reads; (3) exonic nonsynonymous variant effect; and (4) combined annotation-dependent depletion score >20. Altogether, this resulted in the discovery of RPA1 variants reported in this study.
Single-cell DNA and protein sequencing
Single-cell DNA sequencing of BM cells was performed by using a custom targeted panel on the Tapestri Platform (Mission Bio). Briefly, a set of amplicons including RPA1 variants found in P1 (germline c.718G>A, chr17:1782314:G>A and somatic c.1735G>T, chr17:1798378:G>T) were amplified as previously reported34 and outlined in detail in the supplemental Methods (available on the Blood Web site). In addition, we used oligonucleotide-conjugated antibodies targeting CD3, CD11b, CD19, CD34, CD38, CD45RA, and CD90 cell surface proteins.
Additional methods
Additional methods are outlined in detail in the supplement: telomere length assessment (quantitative FISH, Southern blot, Flow-FISH, and telomere shortest length assay), biochemical studies of RPA variants (expression, purification, and Förster resonance energy transfer), iPSC model (CRISPR/Cas9–targeted mutagenesis, quality assessment, hematopoietic differentiation, cytospins, protein expression, irradiation and assessment of DNA damage, and flow cytometry), genomics (deep sequencing, BM-derived single colony sequencing, RPA1 allelic quantification, digital droplet polymerase chain reaction, SNP arrays, haplotype phasing and quantification of uniparental isodisomy, and Sanger sequencing), and statistics.
Results
Heterogeneous clinical manifestations of TBD/STS unified by short telomeres in the study cohort
In the process of identifying new genetic causes of Mendelian disorders, we compiled an international cohort of 4 patients with phenotypes consistent with TBD/STS and yet unidentified genetic cause (Table 1; Figure 1A). All patients’ medical records and evaluations underwent central review and had negative workup for inherited BMF syndromes and genes associated with TBD/STS. Unbiased WES found 3 germline heterozygous missense variant alleles in RPA1 (NM_002945.5) as the most plausible candidates in our patient cohort. Previously unreported in population or disease databases, de novo RPA1 c.718G>A, p.E240K and c.808A>G, p.T270A occurred in P1 and P4, respectively. The RPA1 c.680T>C, p.V227A variant found in P2 and P3 occurs at ultra-low frequency in control populations. P2 father and sister are carriers with no history of hematologic manifestations (clinical workup declined), and P3 family members were not available for evaluation. We did not identify other de novo variants in genes involved in hematopoiesis, telomere homeostasis, or cancer that would be compatible with the observed phenotypes. All de novo variants in P1, P2, and P4 are shown in supplemental Table 1. RPA1 is located on chromosome 17p13.3 and has 17 exons coding for a 70 kDa protein that is expressed in all tissues (Figure 1B, top panel). The three RPA1 variants cluster to DBD-A of RPA1 protein (Figure 1B, bottom panel) and are near ssDNA-binding grooves (supplemental Figure 1A). These variants affect semi-conserved to highly conserved nucleotides (supplemental Figure 1B) with resulting amino acid substitutions predicted as likely deleterious.
Genomic and clinical features of patients with germline heterozygous RPA1 variants
| Patient feature | Germline mutation | Disease feature | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Patient | Sex | Age of onset (y) | RPA1 status | Exon | VAF% (TD) | Population database | CADD | Hematopoietic | Other | Recent status |
| P1 | F | 10 | c.718G>A, p.E240K de novo | 9 | 24%* (58) | Absent | 23.3 | Refractory cytopenia of childhood | Mucocutaneous triad Congenital eye anomaly† | No interventions Stable blood counts at age 28 y |
| P2 | F | 13 | c.680T>C, p.V227A | 8 | 51% (118) | gnomAD v3.1: 76078 genomes (1/152156 alleles) TOPMed freeze 8: 132345 genomes (2/264690 alleles)‡ | 26.9 | MDS with excess blasts | Mild restrictive lung disease Facial dysmorphism§ | HSCT Death at age 14 y (GVHD, infection, pulmonary fibrosis) |
| P3 | F | 58 | 53% (49) | BM not examined CBC normal | Familial pulmonary fibrosis‖ | Progression of pulmonary fibrosis on antifibrotics | ||||
| P4 | F | Birth | c.808A>G, p.T270A de novo | 10 | 41% (51) | Absent | 20.9 | BM not examined Low TRECs Lymphopenia Hypogammaglobulinemia | Prematurity (33 wk) | IgG replacement Stable CBC at age 3 y |
| Patient feature | Germline mutation | Disease feature | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Patient | Sex | Age of onset (y) | RPA1 status | Exon | VAF% (TD) | Population database | CADD | Hematopoietic | Other | Recent status |
| P1 | F | 10 | c.718G>A, p.E240K de novo | 9 | 24%* (58) | Absent | 23.3 | Refractory cytopenia of childhood | Mucocutaneous triad Congenital eye anomaly† | No interventions Stable blood counts at age 28 y |
| P2 | F | 13 | c.680T>C, p.V227A | 8 | 51% (118) | gnomAD v3.1: 76078 genomes (1/152156 alleles) TOPMed freeze 8: 132345 genomes (2/264690 alleles)‡ | 26.9 | MDS with excess blasts | Mild restrictive lung disease Facial dysmorphism§ | HSCT Death at age 14 y (GVHD, infection, pulmonary fibrosis) |
| P3 | F | 58 | 53% (49) | BM not examined CBC normal | Familial pulmonary fibrosis‖ | Progression of pulmonary fibrosis on antifibrotics | ||||
| P4 | F | Birth | c.808A>G, p.T270A de novo | 10 | 41% (51) | Absent | 20.9 | BM not examined Low TRECs Lymphopenia Hypogammaglobulinemia | Prematurity (33 wk) | IgG replacement Stable CBC at age 3 y |
CADD, combined annotation-dependent depletion, pathogenicity threshold of 15; CBC, complete blood count; F, female; IgG, immunoglobulin G; TD, total depth; TRECs, T-cell receptor excision circles.
Confirmed germline in fibroblasts; decreased variant allele frequency (VAF) is due to UPD in blood cells.
Congenital persistent hyperplastic primary vitreous of the left eye requiring enucleation.
gnomAD v3.1 and TOPMed Bravo freeze 8 are population databases with population genome sequencing data.
Bilateral blepharophimosis, epicanthus inversus, eyelid ptosis, and thick eyebrows with high arch found in P2 and carrier father.
Pulmonary fibrosis diagnosed in 2 sisters of P3 (supplemental Figure 2). NM_002945.5 was used for variant nomenclature.

Clinical characteristics of patients with identified RPA1 variants. (A) Germline RPA1 variants identified in 4 pedigrees using exome sequencing. Black filled, open dotted, and open symbols denote affected individuals with heterozygous (het) RPA1 mutations, unaffected carriers, and unaffected family members without mutations, respectively. (B) Top panel: schematic of the RPA1 gene with 17 coding exons illustrated as black lines and untranslated regions shown in red (NM_002945.5). The 3 unique patient variants are located on exons 8, 9, and 10. Bottom panel: Human RPA1 protein illustration with 4 oligonucleotide/oligosaccharide–binding fold domains F, A, B, and C with amino acid boundaries shown below. Oligonucleotide/oligosaccharide–binding folds A, B, and C are ssDNA-binding domains. Red arrows indicate location of 3 RPA1 missense alterations. (C) BM findings in patient cohort (top panel): P1 BM aspirate smears (Wright-Giemsa staining) at time of clinical presentation at age 10 years showing marked reduction in cell content with hypoplasia of all lineages (left image) and megaloblastic maturation of erythroid precursors (middle image). P2 BM infiltrated with myeloblasts (right image) consistent with MDS with excess blasts. Pulmonary findings by chest computed tomography (CT) imaging in patient cohort (middle panel): P2 chest CT scan (left image) during hospitalization showing necrotizing pneumonitis with diffuse ground-glass opacities and large air-filled cavities with differential diagnosis, including pulmonary GVHD, opportunistic infection(s), pulmonary fibrosis, or a combination of the aforementioned conditions. P3 presented at age 58 years with cough and exertional dyspnea with chest CT scan (middle image) findings showing intralobular reticulations with traction bronchiectasis and mild honeycombing, with left asymmetric and basal, subpleural predominance. At 61 years, P3 chest CT scan (right image) showed significant progression of asymmetric lung fibrosis with left predominance and massive basal honeycombing. Mucocutaneous abnormalities in P1 (bottom panel): oral leukoplakia at 19 years of age (top left image) with mild improvement at 25 years of age (top right image), nail dystrophy (bottom left image), and reticular skin pigmentation on the ventral neck (bottom right image). (D) Telomere length analysis by flow cytometry–based FISH was conducted in lymphocytes of P1 (red circles) and P4 (blue circles). P1 telomere length is less than the first percentile at 24 and 27 years of age (red circles). P4 telomere length is at the fifth percentile at 1.75 years of age and less than the first percentile at age 3.33 years (blue circles). All measurements were performed in triplicate, and mean telomere length was calculated in kilobases in relation to the internal control (bovine thymocytes) with known telomere length. A total of 356 healthy controls used for calculation of the first (solid line, bottom), fifth (dashed line, bottom), 95th (dashed line, top), and 99th (solid line, top) percentile curves. (E) TRF analysis in peripheral blood DNA from P2 and family and P3 compared with healthy age–matched control (Ctrl), digested with HinfI and RsaI enzymes followed by separation on 0.7% agarose gel.
Clinical characteristics of patients with identified RPA1 variants. (A) Germline RPA1 variants identified in 4 pedigrees using exome sequencing. Black filled, open dotted, and open symbols denote affected individuals with heterozygous (het) RPA1 mutations, unaffected carriers, and unaffected family members without mutations, respectively. (B) Top panel: schematic of the RPA1 gene with 17 coding exons illustrated as black lines and untranslated regions shown in red (NM_002945.5). The 3 unique patient variants are located on exons 8, 9, and 10. Bottom panel: Human RPA1 protein illustration with 4 oligonucleotide/oligosaccharide–binding fold domains F, A, B, and C with amino acid boundaries shown below. Oligonucleotide/oligosaccharide–binding folds A, B, and C are ssDNA-binding domains. Red arrows indicate location of 3 RPA1 missense alterations. (C) BM findings in patient cohort (top panel): P1 BM aspirate smears (Wright-Giemsa staining) at time of clinical presentation at age 10 years showing marked reduction in cell content with hypoplasia of all lineages (left image) and megaloblastic maturation of erythroid precursors (middle image). P2 BM infiltrated with myeloblasts (right image) consistent with MDS with excess blasts. Pulmonary findings by chest computed tomography (CT) imaging in patient cohort (middle panel): P2 chest CT scan (left image) during hospitalization showing necrotizing pneumonitis with diffuse ground-glass opacities and large air-filled cavities with differential diagnosis, including pulmonary GVHD, opportunistic infection(s), pulmonary fibrosis, or a combination of the aforementioned conditions. P3 presented at age 58 years with cough and exertional dyspnea with chest CT scan (middle image) findings showing intralobular reticulations with traction bronchiectasis and mild honeycombing, with left asymmetric and basal, subpleural predominance. At 61 years, P3 chest CT scan (right image) showed significant progression of asymmetric lung fibrosis with left predominance and massive basal honeycombing. Mucocutaneous abnormalities in P1 (bottom panel): oral leukoplakia at 19 years of age (top left image) with mild improvement at 25 years of age (top right image), nail dystrophy (bottom left image), and reticular skin pigmentation on the ventral neck (bottom right image). (D) Telomere length analysis by flow cytometry–based FISH was conducted in lymphocytes of P1 (red circles) and P4 (blue circles). P1 telomere length is less than the first percentile at 24 and 27 years of age (red circles). P4 telomere length is at the fifth percentile at 1.75 years of age and less than the first percentile at age 3.33 years (blue circles). All measurements were performed in triplicate, and mean telomere length was calculated in kilobases in relation to the internal control (bovine thymocytes) with known telomere length. A total of 356 healthy controls used for calculation of the first (solid line, bottom), fifth (dashed line, bottom), 95th (dashed line, top), and 99th (solid line, top) percentile curves. (E) TRF analysis in peripheral blood DNA from P2 and family and P3 compared with healthy age–matched control (Ctrl), digested with HinfI and RsaI enzymes followed by separation on 0.7% agarose gel.
Our patient cohort had a broad spectrum of clinical presentation and age of onset. P1 presented at age 10 years with pancytopenia, hypoplastic BM (Figure 1C), and history of congenital eye anomaly requiring enucleation as an infant (Table 1). She further developed a classic dyskeratosis congenita–associated mucocutaneous triad during adolescence. However, her clinical course was atypical due to stabilization of blood counts and mucocutaneous features without intervention over 18 years. P2, with preexisting facial dysmorphisms, was diagnosed at 13 years of age with MDS with excess blasts and somatic NRAS c.35G>A, p.G12D mutation at 37% allelic frequency. Mild restriction was noted on pulmonary function testing before receiving a myeloablative allogeneic hematopoietic stem cell transplantation (HSCT). Short-term complications of HSCT included severe skin and liver graft-versus-host disease (GVHD) with development of necrotizing pneumonitis with extensive cavitation secondary to multifactorial causes, including pulmonary GVHD, infections (nocardia pneumonitis and pulmonary aspergillosis), and pulmonary fibrosis at 8 months’ post-HSCT.
This patient died 11 months’ post-HSCT of multi-organ failure in setting of subarachnoid hemorrhage and intractable chronic GVHD. P3 had early hair graying and adult-onset IPF that progressed in her fifth decade of life (Table 1; Figure 1C). Two siblings of P3 were also diagnosed with IPF, but genetic studies to segregate RPA1 mutation were not possible due to death caused by IPF-related complications (supplemental Figure 2). P4 presented at birth with prematurity, failure to thrive, and low T-cell receptor excision circles, triggering further workup that was significant for T- and B-cell lymphopenia and severe hypogammaglobulinemia requiring chronic replacement therapy (supplemental Table 2). Due to clinical features consistent with the disease spectrum of TBD/STS, we assessed telomere length in all patients using various standard approaches depending on specimen availability. P1 and P4 had decreased telomere length in peripheral blood lymphocytes compared with age-matched control subjects (Figure 1D) using flow cytometry-based fluorescence in situ hybridization (FISH).35 Specifically, P1 had telomere length less than the first percentile measured at 2 time points 3 years apart, whereas P4 telomere length decreased to less than the first percentile from the fifth percentile over 1.5 years. Telomere restriction fragment (TRF) analysis on DNA from peripheral blood of P2 and P3 revealed short telomeres in P2 compared with family members, as well as in P3 compared with age-matched healthy control (Figure 1E). To gain further resolution, a telomere shortest length assay (TeSLA) was performed in blood DNA, showing a higher proportion of telomeres <1 kb in P2 and P3 (supplemental Figure 3), consistent with increased frequency of very short telomeres.
Germline RPA1 variants exhibit increased binding to ssDNA and telomeric DNA
To assess the effects of human RPA1 variants on RPA heterotrimer formation and function (Figure 2A), we expressed and purified RPA heterotrimers with wild-type RPA1 (RPAWT) and mutant V227A (RPAV227A), E240K (RPAE240K), and T270A (RPAT270A) proteins (supplemental Figure 4A). All proteins did form heterotrimers and bound ssDNA with high affinity in a Förster resonance energy transfer–based DNA-binding assay using Cy3/Cy5-labeled dT30 ssDNA (Figure 2B). All 3 mutant proteins appear to form an altered complex with ssDNA. Each saturates dT30 ssDNA at a lower stoichiometry than wild type: the binding ratio was 2.2 for RPAWT, 1.3 for RPAV227A, 1.1 for RPAE240K, and 1.6 for RPAT270A (Figure 2B; supplemental Table 3). We next examined RPA binding to dT15, which only interacts with DBD-A and DBD-B of RPA1. Compared with RPAWT, RPAV227A and RPAE240K displayed a twofold and eightfold higher affinity, respectively, whereas RPAT270A had a marginally higher (1.3-fold) binding (Figure 2C; supplemental Table 3). When assessing for binding to 15-mer human telomeric sequence (TTAGGG)2TTA, RPAV227A and RPAE240K had increased binding affinity, whereas RPAT270A had no effect compared with RPAWT (Figure 2D; supplemental Table 3). Next, we examined the kinetics of the telomeric G-quadruplexes melting (Figure 2E-F; supplemental Figure 4B-E; supplemental Table 3). RPAE240K and RPAV227A showed a greater extent and rate of melting of telomeric G-quadruplex sequences vs RPAWT, especially at subsaturating protein concentrations. We conclude that all 3 mutants have a high affinity for DNA, and RPAV227A and RPAE240K exert a higher affinity and telomere unfolding capacity than RPAWT.
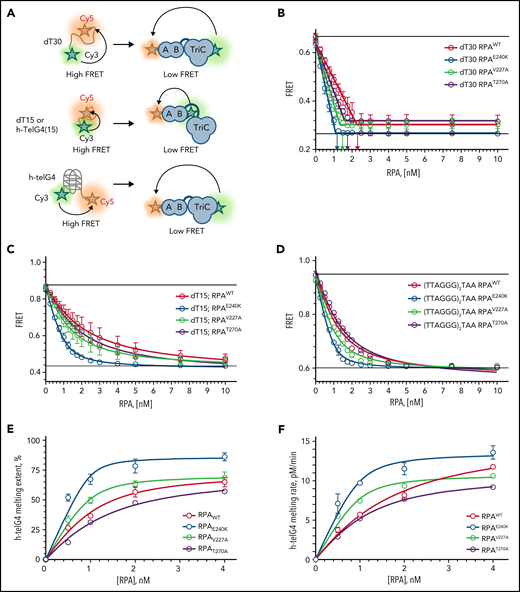
All RPA mutant heterotrimeric proteins exhibit increased affinity for ssDNA, and RPAV227A and RPAE240K possess increased capacity to unfold human telomeric G-quadruplex (h-telG4) DNA. (A) Schematic depiction of the experimental schemes for the Förster resonance energy transfer (FRET)-based assays. Binding of RPA:RPA1WT (RPAWT), RPA:RPA1V227A (RPAV227A), RPA:RPA1V270A (RPAV270A), and RPA:RPA1E240K (RPAE240K) proteins was monitored by using 1 nM dT30 ssDNA molecules (top), 1 nM dT15, or TTAGGGTAAGGGTAA telomeric DNA sequence (middle) labeled with Cy3 and Cy5 fluorescent dyes at the 5′ and 3′ ends, respectively. High FRET corresponds to free ssDNA, and low FRET reflects RPA binding. Unfolding of the h-telG4 was monitored by using the (TTAGGG)5 sequence (bottom). FRET between the Cy3 and Cy5 dyes calculated for the h-telG4 in the absence of proteins and in buffer containing K+ corresponds to 100% folded quadruplex, whereas the FRET value of h-telG4 in the presence of saturating concentrations of RPAE240K in buffer containing Li+ corresponds to 100% unfolded h-telG4. (B) Stoichiometric binding (1 RPA: 1 dT30 molecule) was observed for RPAE240K, and nearly stoichiometric binding was observed for RPAWT, RPAV227A, and RPAV270A. The arrows mark the respective protein concentrations at inflection points of the 2-line linear regression fit. dT15 (C) and TTAGGGTAAGGGTAA (D) telomeric DNA sequence binding to RPAWT, RPAV227A, RPAV270A, and RPAE240K. The data were fitted to a quadratic binding equation. The calculated Kds with respective fitting errors are listed in supplemental Table 3. (E-F) Melting of the h-telG4 DNA, stabilized by the presence of 100 mM KCl. (E) Extent of the h-telG4 melting reactions was calculated from the plateaus of each respective time course. (F) h-telG4 melting rates for RPAWT, RPAV227A, RPAV270A, and RPAE240K were calculated from the slopes of FRET change during the first 20 seconds of each time course (supplemental Figure 4). The data were fitted to a quadratic binding equation. The calculated apparent Kds with respective fitting errors are listed in supplemental Table 3. In all panels, the data are shown as average for 3 independent experiments. Error bars represent standard deviation. Where not shown, error bars are smaller than the data points.
All RPA mutant heterotrimeric proteins exhibit increased affinity for ssDNA, and RPAV227A and RPAE240K possess increased capacity to unfold human telomeric G-quadruplex (h-telG4) DNA. (A) Schematic depiction of the experimental schemes for the Förster resonance energy transfer (FRET)-based assays. Binding of RPA:RPA1WT (RPAWT), RPA:RPA1V227A (RPAV227A), RPA:RPA1V270A (RPAV270A), and RPA:RPA1E240K (RPAE240K) proteins was monitored by using 1 nM dT30 ssDNA molecules (top), 1 nM dT15, or TTAGGGTAAGGGTAA telomeric DNA sequence (middle) labeled with Cy3 and Cy5 fluorescent dyes at the 5′ and 3′ ends, respectively. High FRET corresponds to free ssDNA, and low FRET reflects RPA binding. Unfolding of the h-telG4 was monitored by using the (TTAGGG)5 sequence (bottom). FRET between the Cy3 and Cy5 dyes calculated for the h-telG4 in the absence of proteins and in buffer containing K+ corresponds to 100% folded quadruplex, whereas the FRET value of h-telG4 in the presence of saturating concentrations of RPAE240K in buffer containing Li+ corresponds to 100% unfolded h-telG4. (B) Stoichiometric binding (1 RPA: 1 dT30 molecule) was observed for RPAE240K, and nearly stoichiometric binding was observed for RPAWT, RPAV227A, and RPAV270A. The arrows mark the respective protein concentrations at inflection points of the 2-line linear regression fit. dT15 (C) and TTAGGGTAAGGGTAA (D) telomeric DNA sequence binding to RPAWT, RPAV227A, RPAV270A, and RPAE240K. The data were fitted to a quadratic binding equation. The calculated Kds with respective fitting errors are listed in supplemental Table 3. (E-F) Melting of the h-telG4 DNA, stabilized by the presence of 100 mM KCl. (E) Extent of the h-telG4 melting reactions was calculated from the plateaus of each respective time course. (F) h-telG4 melting rates for RPAWT, RPAV227A, RPAV270A, and RPAE240K were calculated from the slopes of FRET change during the first 20 seconds of each time course (supplemental Figure 4). The data were fitted to a quadratic binding equation. The calculated apparent Kds with respective fitting errors are listed in supplemental Table 3. In all panels, the data are shown as average for 3 independent experiments. Error bars represent standard deviation. Where not shown, error bars are smaller than the data points.
RPA1E240K mutation results in premature telomere shortening and abnormal hematopoiesis in an iPSC model
To understand the effect of RPA1 variants on telomere length regulation and hematopoiesis (Figure 3A), CRISPR/Cas9–mediated mutagenesis was used to introduce a homozygous E240K variant (denoted as RPA1E240K) into the endogenous RPA1 locus of a healthy female donor iPSC line (RPA1WT). RPA1 whole-gene sequencing and Sanger analysis confirmed on-target mutagenesis. RPA1WT and RPA1E240K iPSC lines had similar RPA1 protein expression (Figure 3B), normal karyotype (supplemental Figure 5A), and intact pluripotency (supplemental Figure 5B). Given the role of RPA1 in replication and DNA repair, we assessed baseline cell cycle and activation of DNA damage response to irradiation, which showed similar cell cycle profiles and γ-H2AX signaling in both RPA1WT and RPA1E240K iPSCs (supplemental Figure 6A-B). We then measured telomere length using quantitative FISH (Figure 3C; supplemental Figure 6C) and TRF (supplemental Figure 6D), which revealed significantly shorter telomeres in RPA1E240K compared with RPA1WT iPSC independent of cell passage. Significant telomere length reduction was also observed in RPA1E240K iPSC-derived hematopoietic progenitors (HPs) (Figure 3D). To assess whether RPA1E240K affects hematopoiesis and whether the RPA1E240K iPSC model could faithfully recapitulate the P1 phenotype of pancytopenia, we evaluated iPSC-derived hematopoietic differentiation with flow cytometric enumeration of cell subsets (supplemental Figure 7). Consistent with reduced hematopoietic potential, RPA1E240K iPSC yielded a lower percentage of cells with the CD43+CD45+ hematopoietic phenotype compared with RPA1WT (Figure 3E). Differentiation of HP cells (CD34+CD43+) to terminal lineages was also compromised. Specifically, RPA1E240K produced significantly less CD71+CD235+ erythroid (Figure 3F) and CD45+CD18+CD11b+ myeloid (Figure 3G) populations with cytologic evidence of scarce orthochromatic proerythroblasts (supplemental Figure 8A-B) and large macrophage-like cells, respectively (supplemental Figure 8C-D).
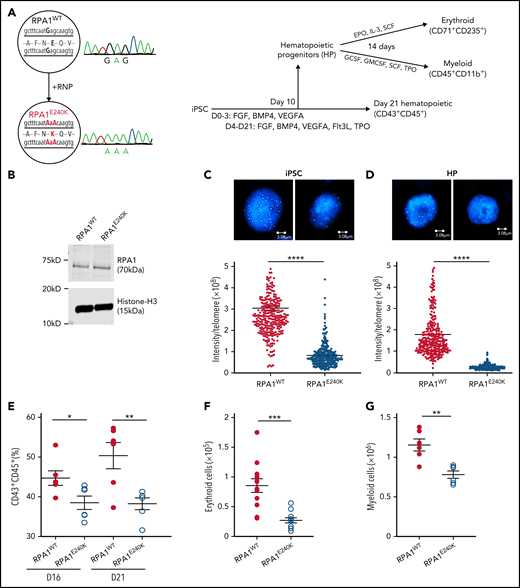
Human RPA1E240K iPSC exhibit telomere shortening and reduced hematopoietic potential. (A) Left panel: Healthy control iPSC (RPA1WT) following CRISPR/Cas9–guided homozygous c.718G>A (p.E240K, as E240K) modification within endogenous RPA1 locus (RPA1E240K) confirmed with Sanger analysis. Right panel: Illustration of iPSC monolayer-based differentiation to HPs and subsequently to erythroid and myeloid cell lineages. Day 10 HP cells were fluorescence-activated cell-sorted and cultured in erythroid or myeloid differentiation media for 14 days. In parallel, iPSC-derived HP were further cultured until day 21 to assess for expression of pan-hematopoietic markers. (B) Immunoblot analysis of RPA1 expression in RPA1WT and RPA1E240K iPSC whole-cell extracts with histone H3 as loading control. Telomere length in RPA1WT passage 17 and RPA1E240K passage 12 iPSCs (C) and iPSC-derived HP cells (D) using quantitative FISH. Graphs represent mean ± standard error of the mean (SEM) of 1 of 3 independent experiments (Student t test, ****P < .0001). (E) Decreased percentage of CD43+CD45+RPA1E240K hematopoietic cells compared with RPA1WT at days 16 and 21. Data represent mean ± SEM of 2 independent experiments (Student t test, *P = .03, **P = .0074). (F) Graphical representation of CD71+CD235+ erythroid cells from iPSC-derived erythroid cultures at day 14. Data represent mean ± SEM of 4 independent experiments (Student t test, ***P = .0002). (G) Plot representation of CD45+CD11b+ myeloid cells from iPSC cultures. Data represent mean ± SEM of 2 independent experiments (Student t test, **P = .0018). EPO, erythropoietin; IL-3, interleukin-3; GMCSF, granulocyte-macrophage colony-stimulating factor; GCSF, granulocyte colony-stimulating factor; SCF, stem cell factor; TPO, thrombopoietin.
Human RPA1E240K iPSC exhibit telomere shortening and reduced hematopoietic potential. (A) Left panel: Healthy control iPSC (RPA1WT) following CRISPR/Cas9–guided homozygous c.718G>A (p.E240K, as E240K) modification within endogenous RPA1 locus (RPA1E240K) confirmed with Sanger analysis. Right panel: Illustration of iPSC monolayer-based differentiation to HPs and subsequently to erythroid and myeloid cell lineages. Day 10 HP cells were fluorescence-activated cell-sorted and cultured in erythroid or myeloid differentiation media for 14 days. In parallel, iPSC-derived HP were further cultured until day 21 to assess for expression of pan-hematopoietic markers. (B) Immunoblot analysis of RPA1 expression in RPA1WT and RPA1E240K iPSC whole-cell extracts with histone H3 as loading control. Telomere length in RPA1WT passage 17 and RPA1E240K passage 12 iPSCs (C) and iPSC-derived HP cells (D) using quantitative FISH. Graphs represent mean ± standard error of the mean (SEM) of 1 of 3 independent experiments (Student t test, ****P < .0001). (E) Decreased percentage of CD43+CD45+RPA1E240K hematopoietic cells compared with RPA1WT at days 16 and 21. Data represent mean ± SEM of 2 independent experiments (Student t test, *P = .03, **P = .0074). (F) Graphical representation of CD71+CD235+ erythroid cells from iPSC-derived erythroid cultures at day 14. Data represent mean ± SEM of 4 independent experiments (Student t test, ***P = .0002). (G) Plot representation of CD45+CD11b+ myeloid cells from iPSC cultures. Data represent mean ± SEM of 2 independent experiments (Student t test, **P = .0018). EPO, erythropoietin; IL-3, interleukin-3; GMCSF, granulocyte-macrophage colony-stimulating factor; GCSF, granulocyte colony-stimulating factor; SCF, stem cell factor; TPO, thrombopoietin.
Somatic inactivation of germline RPA1 mutation results in benign clonal hematopoiesis with long-term potential
The atypical clinical course in P1 with stable blood counts over 2 decades (Figure 4A) without progression of leukoplakia (Figure 1C) and telomere shortening (Figure 1D) led us to examine patient specimens for potential rescue mechanisms. We found that allelic frequency of the RPA1 c.718G>A, p.E240K variant is reduced in BM (27%) compared with fibroblast DNA (50%) (Figure 4B). Two somatic events were identified in BM: a second-site truncating (stop-gain) RPA1 mutation c.1735G>T, p.K579* at 10% allelic frequency and a uniparental isodisomy of chromosome 17p (UPD17p) (Figure 4C). To understand the mechanism of these rescue events, we first confirmed the stop-gain c.1735G>T mutation to be in cis with germline c.718G>A allele (Figure 4D; supplemental Figure 9). Bulk RNA sequencing of BM cells revealed total loss of the somatic mutation concurrent with reduction of the germline variant to 13%. Ultra-deep RNA sequencing performed to accurately quantify these mutations confirmed nearly absent expression (0.8%) of somatic c.1735G>T substitution (Figure 4E). To assess whether the 2 mosaic events were sustained over time, we performed serial single nucleotide polymorphism (SNP) array and deep sequencing of hematopoietic cells. We also found a significant expansion of UPD17p clone over 15 years, which corresponds to the declining germline c.718G>A allele on serial BM evaluations (Figure 4F). In addition, we observed an independent increase in clonal burden of somatic c.1735G>T, p.K579* mutation over time.
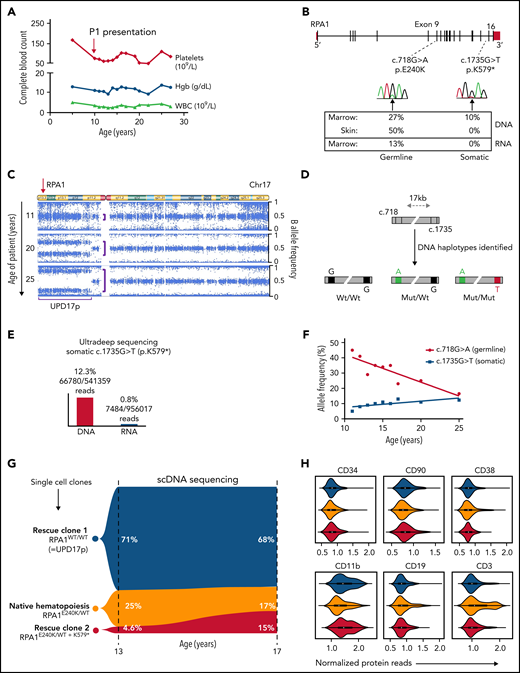
Natural evolution of disease and somatic genetic rescue in P1. (A) White blood cells (WBC, triangles), hemoglobin (Hgb, circles), and platelets (diamonds) plotted over 23 years for P1 (pedigree 1). Red arrow indicates time of clinical presentation. (B) Illustration of germline RPA1 variant in exon 9 and somatic mutation in exon 16 with respective DNA Sanger electropherograms from BM. Bottom table depicts variant allelic frequencies from exome sequencing performed in BM and skin fibroblast DNA and RNA sequencing from BM when patient was aged 20 years. (C) Copy number neutral UPD encompassing RPA1 locus at 17p13.3 (red arrow) identified by using an SNP array. Serial SNP array analysis in BM granulocytes shows UPD expansion over time (denoted by purple brackets). (D) Schematic of RPA1 locus (gray bar) with germline (c.718) and somatic (c.1735) mutational spots 17 kb apart. Three haplotype orientations between c.718 and c.1735 identified in marrow DNA of P1 at age 19 years from 2 independent experiments using digital droplet polymerase chain reaction: left haplotype, wt/wt (c.718G wild type/c.1735G wild type) denoted by black boxes; middle haplotype, mut/wt (c.718A mutant/c.1735G wild type) denoted by green and black boxes; and right haplotype, mut/mut (c.718A mutant/c.1735T mutant) denoted by green and red boxes. (E) Ultra-deep amplicon sequencing of bone marrow DNA and RNA targeting position of RPA1 somatic mutation (c.1735) confirms near total loss of mutant RNA. (F) Longitudinal deep sequencing in BM samples from diagnosis to age 25 years showing decrease in allele frequency of the germline c.G718G>A variant (red line) and increase of the somatic c.1735G>T mutation (blue line). (G) Single cells from P1 BM at ages 13 and 17 years were sequenced for germline (RPA1:chr17:1782314:G>A) and somatic (RPA1:chr17:1798378:G>T) mutational positions using single-cell DNA (scDNA) sequencing Tapestri Platform. Violin plot shows 3 clonal populations, including homozygous wild type (blue, RPA1WT/WT; rescue clone 1 = UPD17p), heterozygous RPA1E240K/WT (gold, native state hematopoiesis), and heterozygous c.718G>A with concurrent c.1735G>T stop-gain (red, RPA1E240K/WT + K579*= rescue clone 2). (H) Tapestri single-cell multi-omic analysis combining DNA mutation data and surface protein expression performed in P1 BM at age 17 years. Panels depict 3 clones (color coding identical to that in panel G) constructed from 2110 high-quality cells with normalized protein expression of markers for hematopoietic stem and progenitor cells (CD34), stem cells (CD90), progenitors (CD38), and terminally differentiated cells, including myeloid (CD11b), B-lymphoid (CD19), and T-lymphoid (CD3) cells.
Natural evolution of disease and somatic genetic rescue in P1. (A) White blood cells (WBC, triangles), hemoglobin (Hgb, circles), and platelets (diamonds) plotted over 23 years for P1 (pedigree 1). Red arrow indicates time of clinical presentation. (B) Illustration of germline RPA1 variant in exon 9 and somatic mutation in exon 16 with respective DNA Sanger electropherograms from BM. Bottom table depicts variant allelic frequencies from exome sequencing performed in BM and skin fibroblast DNA and RNA sequencing from BM when patient was aged 20 years. (C) Copy number neutral UPD encompassing RPA1 locus at 17p13.3 (red arrow) identified by using an SNP array. Serial SNP array analysis in BM granulocytes shows UPD expansion over time (denoted by purple brackets). (D) Schematic of RPA1 locus (gray bar) with germline (c.718) and somatic (c.1735) mutational spots 17 kb apart. Three haplotype orientations between c.718 and c.1735 identified in marrow DNA of P1 at age 19 years from 2 independent experiments using digital droplet polymerase chain reaction: left haplotype, wt/wt (c.718G wild type/c.1735G wild type) denoted by black boxes; middle haplotype, mut/wt (c.718A mutant/c.1735G wild type) denoted by green and black boxes; and right haplotype, mut/mut (c.718A mutant/c.1735T mutant) denoted by green and red boxes. (E) Ultra-deep amplicon sequencing of bone marrow DNA and RNA targeting position of RPA1 somatic mutation (c.1735) confirms near total loss of mutant RNA. (F) Longitudinal deep sequencing in BM samples from diagnosis to age 25 years showing decrease in allele frequency of the germline c.G718G>A variant (red line) and increase of the somatic c.1735G>T mutation (blue line). (G) Single cells from P1 BM at ages 13 and 17 years were sequenced for germline (RPA1:chr17:1782314:G>A) and somatic (RPA1:chr17:1798378:G>T) mutational positions using single-cell DNA (scDNA) sequencing Tapestri Platform. Violin plot shows 3 clonal populations, including homozygous wild type (blue, RPA1WT/WT; rescue clone 1 = UPD17p), heterozygous RPA1E240K/WT (gold, native state hematopoiesis), and heterozygous c.718G>A with concurrent c.1735G>T stop-gain (red, RPA1E240K/WT + K579*= rescue clone 2). (H) Tapestri single-cell multi-omic analysis combining DNA mutation data and surface protein expression performed in P1 BM at age 17 years. Panels depict 3 clones (color coding identical to that in panel G) constructed from 2110 high-quality cells with normalized protein expression of markers for hematopoietic stem and progenitor cells (CD34), stem cells (CD90), progenitors (CD38), and terminally differentiated cells, including myeloid (CD11b), B-lymphoid (CD19), and T-lymphoid (CD3) cells.
Analysis of clonality at single-cell resolution identifies mutually exclusive rescue events that arise in early hematopoiesis
To dissect the clonal architecture at the single-cell level, we interrogated P1 BM using single-cell DNA sequencing. Three clones with unique RPA1 allelic patterns were detected at age 13 years: heterozygous c.718G>A (native state hematopoiesis), homozygous wild type (rescue clone 1 with UPD17p), and heterozygous c.718G>A with concurrent somatic c.1735G>T (rescue clone 2 with stop-gain mutation) (Figure 4G). This clonal pattern was confirmed in HP colonies derived from BM (supplemental Figure 10A). Clonal trajectories assessed over 4 years exhibited minor expansion of rescue clone 2 at the expense of native hematopoiesis. Finally, we investigated clonal origins within the hematopoietic hierarchy by using barcoded antibodies in the same reaction to track surface markers on the single-cell level. Both rescue clones were found within hematopoietic stem and progenitor cells characterized by the expression of CD34, CD90, and CD38 (Figure 4H). In addition, compared with native hematopoiesis, these clones were enriched in CD11b+ myeloid and CD19+ B cells but markedly reduced in CD3+ T lymphocytes. Similar skewing of UPD17p lesion toward myeloid lineage was observed comparing SNP array results in bulk granulocytes vs lymphocytes (supplemental Figure 10B). The enrichment of both somatic clones in CD11+ and CD19+ cells with a near absence of rescue hematopoiesis in T lymphocytes is likely due to their long life span compared with short-lived myeloid and B-cell lineages. Of note, no malignant somatic alterations (ie, clonal hematopoiesis of indeterminate potential) were found by using WES in BM after diagnosis.
Discussion
It was not until the late 20th century that DKC1 was identified as the first gene to cause dyskeratosis congenita, a syndrome described in 1910 with features of nail dystrophy, oral leukoplakia, and skin pigmentation anomalies.36 Since then, an increasing number of genes involved in telomere homeostasis have been associated with classical phenotypes of TBD/STS that include BMF and the mucocutaneous triad but also manifestations such as isolated pulmonary fibrosis. Our study expands the genetic spectrum of this entity by describing 3 germline RPA1 variants clustering to DBD-A as associated with a novel Mendelian disorder that clinically resembles a TBD/STS with short telomeres. Consistent with the variable expressivity and penetrance observed in TBD/STS, the 4 affected individuals developed a wide range of disease features at various ages with the common denominator of telomere shortening. Constitutional manifestations included facial and eye anomalies present in 2 patients, pulmonary disease in 2, and a classical mucocutaneous triad in 1. The hematopoietic system was affected in 3 patients who had BMF, MDS, T- and B-cell lymphopenia, and hypogammaglobulinemia. Only one patient (P3) did not have abnormal blood counts, and thus no further marrow analysis was needed. Similar to other manifestations of TBD/STS reported in the literature,37 it is possible that this patient’s disease had slow progression in the hematopoietic system while resulting in premature aging in the lungs manifesting as pulmonary fibrosis.
The well-established role of RPA1 in telomere maintenance using mammalian and non-mammalian model systems predated our discovery of germline RPA1 mutations in patients with TBD/STS and short telomeres. Historically, many Saccharomyces cerevisiae rfa1 (RPA1 paralog) mutants exhibited increased sensitivity from DNA damage, defective checkpoints, and gross chromosomal aberrations owing to RPA1 function in DNA replication and repair.18,19,38 Smith et al38 first showed the presence of RPA at the telomeric ends maximally during S phase, as well as telomere shortening in a synergistic yku70-rfa1-D228Y S cerevisiae model. rfa1-D228Y was then modeled in Schizosaccharomyces pombe (paralog is rad11-D223Y), which confirmed sensitivity to UV and γ-irradiation and exhibited reduced telomere length, suggesting that RPA is directly involved in telomere maintenance.32 Follow-up studies further supported the role of RPA1 in telomere biology by showing the ability of RPA1 to bind and unfold telomeric G-quadruplexes,26,28,39 regulate telomerase and telomerase access to chromosomal ends,24,40 and prevent accumulation of telomeric ssDNA in cells positive for alternative lengthening of telomeres.29 Furthermore, rad11-D223Y in S pombe, corresponding to human RPA1 D228Y, was shown to reduce binding affinity for telomeric ssDNA and G-quadruplexes, resulting in telomere shortening.25-27 Overall, multiple studies have established the role of RPA1 in telomere biology.
Unlike the previously studied variants, all patient RPA1 mutant proteins had preserved binding to ssDNA. Furthermore, RPA1V227A and RPA1E240K harbored significantly enhanced binding to ssDNA and telomeric sequences and a greater rate and extent of melting of G-quadruplexes, whereas the RPA1T270A mutant was equivocal to RPA1WT. Binding of RPA to DNA is dynamic and regulates loading of other essential proteins on ssDNA. It is plausible that GOF RPA1V227A and RPA1E240K mutations alter the access between chromosomal ends and telomere maintenance machinery such as shelterin or telomerase complex and/or challenge the RPA-to-POT1 switch and POT1 capping function, ultimately leaving telomeric ends vulnerable to telomere degrading transactions.41 In addition, specific point mutations might affect interactions of RPA1 with other proteins, which could explain why RPA1T270A behaves differently from other RPA1 mutants in binding to telomere sequences. The molecular mechanism by which RPA1 exerts telomere maintenance is highly complex and remains elusive, requiring further investigation.
To directly examine whether a single amino acid exchange in RPA1 can be deleterious to eukaryotic cells, we modeled the RPA1E240K variant in an iPSC model derived from a healthy donor. We deliberately chose to introduce patient RPA1E240K mutation into a healthy donor iPSC cell line instead of generating patient-derived iPSCs to eliminate the confounding effect of other unknown patient-specific mutations. The rationale for establishing a homozygous knock-in was to avoid the development of rescue events found in P1 and to exaggerate the biological phenotype for a gene variant that is associated with a late-onset disease. We were able to recapitulate the biological phenotype of significant telomere shortening in RPA1E240K iPSC, as well as iPSC-derived hematopoiesis. Finally, we showed that the RPA1E240K mutation resulted in reduced capacity to generate iPSC-derived HPs and decreased erythroid/myeloid differentiation. This is in line with other reported iPSC short telomere disease models in which hematopoietic insufficiency has been observed.42
Stereotypical dyskeratosis congenita displays a progressive course with BMF leading to severe cytopenias over time.6 Remarkably, over 18 years, P1 followed an atypical course for TBD/STS marked by stabilization of hematologic features. Using this thread, we unraveled the unique propensity of the germline RPA1E240K mutation to facilitate the development of 2 independent somatic genetic escape lesions in P1. These were a second-site truncating RPA1 mutation causing degradation of germline mutant RNA and an UPD17p recombination resulting in replacement of germline variant with a wild-type allele. It seems that these mosaic events can expand and outcompete the native state RPA1-mutated hematopoiesis with no signs of exhaustion or malignant transformation. Somatic genetic rescue in hematopoiesis has been described in TBD/STS genes such as TERC, TERT, NHP2, TINF2, and DKC1, which arise in response to loss-of-function mutations.43-49 The novelty of our finding lies in the identification of somatic rescue arising in response to a GOF RPA1E240K mutation, a known phenomenon described in patients with GOF mutations in SAMD9/SAMD9L genes.34,50-52 We also observed improvement of oral leukoplakia in P1 over time. One can speculate that mucosal tissue in this patient also underwent somatic reversion, given that somatic mosaicism has been recently shown to be common in healthy human tissues.53
Our study does have potential limitations that should be noted. First, due to the small size of our cohort, we were unable to characterize the full phenotypic spectrum associated with RPA1 mutations. Second, telomere length was measured either by flow FISH or TRF Southern blot, which was inherent to our patient cohort, presenting in 4 countries at different ages to unique providers with different strategies for sample banking. Although outside the scope of this study, further research efforts are required to understand the exact mechanism of how RPA1 mutations cause telomere shortening, how RPA regulates telomere length, and how this compares with other telomere-associated genes.54,55
In summary, we identified RPA1 mutations as associated with telomere shortening in humans, which calls for careful consideration of RPA1 missense variants in the workup of patients with phenotypes of TBD/STS. Germline RPA1 variants can either be permissive, as seen with RPA1V227A that is associated with reduced penetrance, or severely “hematotoxic” leading to somatic inactivation, as observed with RPA1E240K. We speculate that germline RPA1 alterations may be more common in human disease, given that somatic RPA1 mutations occur in ∼1% of cancers.56 Additional efforts are needed to not only determine further pathogenic RPA1 variants but to also elucidate the role of RPA1 in human telomere biology.
Acknowledgments
The authors thank the patients for participation. The authors also acknowledge their collaborators: Dirk Lebrecht, Marco Teller, Ali-Riza Kaya, Wilfried Truckenmüller, Maria Siskou-Zwecker, and Axel Gebert (Freiburg, Germany) for laboratory assistance and data management; Loizos Petrikkos and Kondylia Antoniadi (Athens, Greece) for patient management; Ibrahima Ba for technical assistance and Bruno Crestani for helpful discussions (Paris, France); Lindsay Burrage for exome analysis and Filiz Seeborg for P4 referral; Yawei Hui, Shibiao Wan, Yiping Fan, and Gang Wu (St Jude Center for Applied Bioinformatics); OMICS computing cluster (University of Lübeck); Emmanuelle Olivier and Patrick Nitschké (Imagine Institute/Université de Paris) for bioinformatics support; Robert Durruthy-Durruthy (Mission Bio) for support on single-cell DNA sequencing analysis; Amabel Orogo (Illumina) and Jordan Sheetz (Bio-Rad Laboratories) for technical support; Mihaela Onciu (Department of Pathology, St Jude Children’s Research Hospital) for iPSC-derived erythroid and myeloid cytology review; Aaron Taylor (St Jude Center of Imaging) for imaging consultation; Sunita Dsouza, Maria Lillo Osuna, and Min-Joon Han (St Jude Children’s Research Hospital) for iPSC technical assistance; Virginia Valentine and Julia Wilbourne (St Jude Cytogenetics Core); and Mitchell Weiss and John Crispino for helpful discussions.
This work was supported by grants from the ERAPERMED GATA2-HuMo 2018-123, Deutsche Krebshilfe Max Eder Grant 109005, Fritz-Thyssen Foundation 10.17.1.026MN, and St Jude American Lebanese Syrian Associated Charities (M.W.W.), José Carreras Leukämie-Stiftung (V.P.L.), BMBF MyPred 01GM1911A (C.M.N., M.W.W., and M.E.). Research reported in the manuscript was supported by the National Institutes of Health (NIH) Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under Award U01HG007709. The St Jude Cancer Center Core Cytogenetics Laboratory is supported by the NIH, National Cancer Institute (P30 CA21765-41), and American Lebanese Syrian Associated Charities. M.S. is supported by the NIH, National Institute of General Medical Sciences (R35GM131704), and the National Cancer Institute (P30CA086862). C.K., C.S., P.R., and S.C. are supported by the Agence Nationale de la Recherche (ANR-20-CE12-0012TeloRPA). H.B. and A.K. are supported by Deutsche Forschungsgemeinschaft (German Research Foundation) under Germany’s Excellence Strategy (EXC 22167-390884018). The P3 exome study was supported by a grant from the Chancellerie des Universités de Paris (legs Poix). The laboratories of V.G. and P.R. are supported by the “Ligue Nationale Contre le Cancer” (Equipe Laboratoryélisée). P.R. is a scientist from Centre National de la Recherche Scientifique. S.C. is supported by Project Fondation ARC, Projet Emergence-Cancéropôle PACA. This work was generated within the European Reference Network for Paediatric Cancer (PAEDCAN). The authors acknowledge the contribution of the Center of Inborn and Acquired Blood Diseases at the Freiburg Center for Rare Diseases, and the Hilda Biobank at the Department of Pediatrics and Adolescent Medicine, Freiburg, Germany. M.B. is supported by the Deutsche Forschungsgemeinschaft (DFG) - CRC 1479 (Project ID: 441891347-S1), CRC 1160 (Project Z02), CRC1453 (Project ID 431984000-S1) and T RR167 (Project Z01), the German Federal Ministry of Education and Research by MIRACUM within the Medical Informatics Funding Scheme (FKZ 01ZZ1801B).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Authorship
Contribution: R.S., S.S.S., M.W.W., C.S., S.C., M.S.W., and M.S. conceived and designed the experiments; R.S., S.S.S., A.K., H.B., T.-C.C., M.B., V.P.L., J.A.R., Ch.K., and M.W.W. performed genomic data analysis; R.S., M.H., S.L.G., C.G., L.S., F.B., M.B.V., S.M.P.-M., A.G.F., D.C., V.G., C.S., P.R., M.S.W., M.S., and S.C. performed and/or interpreted functional experiments; F.B., S.H., M.L., Ch.K., M.A.C., S.N., J.A.R., S.P., C.M.N., M.E., R.S., and M.W.W. were involved in patient care, collecting clinical data, and clinical testing; M.W.W., S.C., M.S.W., and M.S. supervised the experiments; M.W.W. oversaw study design; and all authors contributed to the manuscript and approved of the final version.
A complete list of the members of the Undiagnosed Diseases Network appears in the Appendix.
Conflict-of-interest disclosure: The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing completed at Baylor Genetics Laboratories. The authors declare no competing financial interests.
Correspondence: Marcin W. Wlodarski, St. Jude Children’s Research Hospital, 262 Danny Thomas Place, MS 341, Memphis, TN 38105; e-mail: marcin.wlodarski@stjude.org.
Python-based Mosaic package for analysis of single-cell DNA sequencing is available at GitHub (https://github.com/MissionBio/mosaic). Raw data sets of WES and single-cell DNA sequencing have been deposited at the European Genome-Phenome Archive (EGA; http://www.ebi.ac.uk/ega/) hosted by the European Bioinformatics Institute under accession numbers EGAS00001005761 (WES) and EGAS00001005762 (single-cell DNA sequencing and cell surface protein analysis).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Appendix: study group members
The members of the Undiagnosed Diseases Network are Maria T. Acosta, Margaret Adam, David R. Adams, Pankaj B. Agrawal, Justin Alvey, Laura Amendola, Ashley Andrews, Euan A. Ashley, Mahshid S. Azamian, Carlos A. Bacino, Guney Bademci, Eva Baker, Ashok Balasubramanyam, Dustin Baldridge, Jim Bale, Michael Bamshad, Deborah Barbouth, Pinar Bayrak-Toydemir, Anita Beck, Alan H. Beggs, Edward Behrens, Gill Bejerano, Jimmy Bennet, Beverly Berg-Rood, Jonathan A. Bernstein, Gerard T. Berry, Anna Bican, Stephanie Bivona, Elizabeth Blue, John Bohnsack, Carsten Bonnenmann, Devon Bonner, Lorenzo Botto, Brenna Boyd, Lauren C. Briere, Elly Brokamp, Gabrielle Brown, Elizabeth A. Burke, Lindsay C. Burrage, Manish J. Butte, Peter Byers, William E. Byrd, John Carey, Olveen Carrasquillo, Ta Chen Peter Chang, Sirisak Chanprasert, Hsiao-Tuan Chao, Gary D. Clark, Terra R. Coakley, Laurel A. Cobban, Joy D. Cogan, Matthew Coggins, F. Sessions Cole, Heather A. Colley, Cynthia M. Cooper, Heidi Cope, William J. Craigen, Andrew B. Crouse, Michael Cunningham, Precilla D’Souza, Hongzheng Dai, Surendra Dasari, Joie Davis, Jyoti G. Dayal, Matthew Deardorff, Esteban C. Dell’Angelica, Katrina Dipple, Daniel Doherty, Naghmeh Dorrani, Argenia L. Doss, Emilie D. Douine, David D. Draper, Laura Duncan, Dawn Earl, David J. Eckstein, Lisa T. Emrick, Christine M. Eng, Cecilia Esteves, Marni Falk, Liliana Fernandez, Carlos Ferreira, Elizabeth L. Fieg, Laurie C. Findley, Paul G. Fisher, Brent L. Fogel, Irman Forghani, William A. Gahl, Ian Glass, Bernadette Gochuico, Rena A. Godfrey, Katie Golden-Grant, Madison P. Goldrich, David B. Goldstein, Alana Grajewski, Catherine A. Groden, Irma Gutierrez, Sihoun Hahn, Rizwan Hamid, Kelly Hassey, Nichole Hayes, Frances High, Anne Hing, Fuki M. Hisama, Ingrid A. Holm, Jason Hom, Martha Horike-Pyne, Alden Huang, Yong Huang, Laryssa Huryn, Rosario Isasi, Fariha Jamal, Gail P. Jarvik, Jeffrey Jarvik, Suman Jayadev, Lefkothea Karaviti, Jennifer Kennedy, Shamika Ketkar, Dana Kiley, Shilpa N. Kobren, Isaac S. Kohane, Jennefer N. Kohler, Deborah Krakow, Donna M. Krasnewich, Elijah Kravets, Susan Korrick, Mary Koziura, Joel B. Krier, Seema R. Lalani, Byron Lam, Christina Lam, Grace L. LaMoure, Brendan C. Lanpher, Ian R. Lanza, Lea Latham, Kimberly LeBlanc, Brendan H. Lee, Hane Lee, Roy Levitt, Richard A. Lewis, Sharyn A. Lincoln, Pengfei Liu, Xue Zhong Liu, Nicola Longo, Sandra K. Loo, Joseph Loscalzo, Richard L. Maas, John MacDowall, Ellen F. Macnamara, Calum A. MacRae, Valerie V. Maduro, Bryan C. Mak, May Christine V. Malicdan, Laura A. Mamounas, Teri A. Manolio, Rong Mao, Kenneth Maravilla, Thomas C. Markello, Ronit Marom, Gabor Marth, Beth A. Martin, Martin G. Martin, Julian A. Martínez-Agosto, Shruti Marwaha, Jacob McCauley, Allyn McConkie-Rosell, Alexa T. McCray, Elisabeth McGee, Heather Mefford, J. Lawrence Merritt, Matthew Might, Ghayda Mirzaa, Eva Morava, Paolo M. Moretti, Deborah Mosbrook-Davis, John J. Mulvihill, Mariko Nakano-Okuno, Avi Nath, Stan F. Nelson, John H. Newman, Sarah K. Nicholas, Deborah Nickerson, Shirley Nieves-Rodriguez, Donna Novacic, Devin Oglesbee, James P. Orengo, Laura Pace, Stephen Pak, J. Carl Pallais, Christina G.S. Palmer, Jeanette C. Papp, Neil H. Parker, John A. Phillips III, Jennifer E. Posey, Lorraine Potocki, Bradley Power, Barbara N. Pusey, Aaron Quinlan, Wendy Raskind, Archana N. Raja, Deepak A. Rao, Genecee Renteria, Chloe M. Reuter, Lynette Rives, Amy K. Robertson, Lance H. Rodan, Jill A. Rosenfeld, Natalie Rosenwasser, Francis Rossignol, Maura Ruzhnikov, Ralph Sacco, Jacinda B. Sampson, Mario Saporta, C. Ron Scott, Judy Schaechter, Timothy Schedl, Kelly Schoch, Daryl A. Scott, Vandana Shashi, Jimann Shin, Rebecca Signer, Edwin K. Silverman, Janet S. Sinsheimer, Kathy Sisco, Edward C. Smith, Kevin S. Smith, Emily Solem, Lilianna Solnica-Krezel, Ben Solomon, Rebecca C. Spillmann, Joan M. Stoler, Jennifer A. Sullivan, Kathleen Sullivan, Angela Sun, Shirley Sutton, David A. Sweetser, Virginia Sybert, Holly K. Tabor, Amelia L.M. Tan, Queenie K.-G. Tan, Mustafa Tekin, Fred Telischi, Willa Thorson, Audrey Thurm, Cynthia J. Tifft, Camilo Toro, Alyssa A. Tran, Brianna M. Tucker, Tiina K. Urv, Adeline Vanderver, Matt Velinder, Dave Viskochil, Tiphanie P. Vogel, Colleen E. Wahl, Stephanie Wallace, Nicole M. Walley, Chris A. Walsh, Melissa Walker, Jennifer Wambach, Jijun Wan, Lee-kai Wang, Michael F. Wangler, Patricia A. Ward, Daniel Wegner, Monika Weisz-Hubshman, Mark Wener, Tara Wenger, Katherine Wesseling Perry, Monte Westerfield, Matthew T. Wheeler, Jordan Whitlock, Lynne A. Wolfe, Jeremy D. Woods, Kim Worley, Shinya Yamamoto, John Yang, Muhammad Yousef, Diane B. Zastrow, Wadih Zein, Chunli Zhao, Stephan Zuchner, Hugo Bellen, and Rachel Mahoney.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal