

In this issue of Blood, perform a comprehensive molecular characterization of a large cohort of >300 patients with a documented diagnosis of splenic marginal zone lymphoma (SMZL).1 By combining genomic and transcriptomic analyses, the authors provide a novel classification of the disease.
SMZL is a mature B-cell neoplasm that accounts for <2% of all non-Hodgkin lym phomas. Clinically, SMZL almost always presents with splenomegaly. Diagnosis can be made using bone marrow biopsy together with phenotypic analysis of circulating B cells. SMZL is an indolent disease, with a median overall survival of ∼10 years. It is usually treated by splenectomy or, more recently, with rituximab.
In this paper, the authors assembled the largest cohort of SMZL cases to date, with 373 patients coming from 28 different centers. All patients entering the study had undergone splenectomy, with tissue available for histological review and none had received treatment before splenectomy. Splenic tissue from 303 eligible patients was analyzed using a CapSeq assay covering a panel of >900 genes that have been reported to be recurrently mutated in SMZL, with a coverage >2000×. Using this approach, NOTCH2, KLF2, and NF-kB were identified as the most frequently mutated genes, confirming previous observations.2-5 An algorithm was then applied to classify these mutations into functional clusters. A minimal set of 14 genes was necessary and sufficient to classify patients in 4 different clusters, namely the NNK cluster, which stands for NF-kB, NOTCH, and KLF2 (accounting for 58.2% of cases), and the DMT cluster, which stands for DNA damage response, MAPK, and TLR (accounting for 32.8% of cases). Smaller clusters (PA [PI3K/AKT] and CBS [cytokine/BCR/spliceosome]) account for the remaining 9% of cases (see figure).
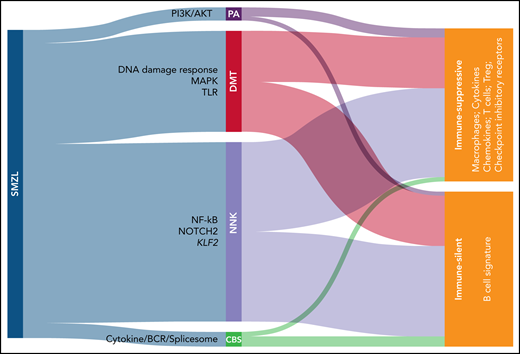
Novel classification of SMZL. Mutational analysis allows to classify SMZL in 4 clusters: PA (PI3K/AKT), DMT (DNA damage response/MAPK/Toll-like receptor), NNK (NF-kB/NOTCH/KLF2), and CBS (cytokine/BCR/spliceosome). SMZLs can be further distinguished based on transcriptome analyses of spleen sections in immune-silent, which express mostly B-associated genes, or immune-suppressive, which express also T-associated genes coding for immunomodulatory molecules.
Novel classification of SMZL. Mutational analysis allows to classify SMZL in 4 clusters: PA (PI3K/AKT), DMT (DNA damage response/MAPK/Toll-like receptor), NNK (NF-kB/NOTCH/KLF2), and CBS (cytokine/BCR/spliceosome). SMZLs can be further distinguished based on transcriptome analyses of spleen sections in immune-silent, which express mostly B-associated genes, or immune-suppressive, which express also T-associated genes coding for immunomodulatory molecules.
Using this set of genes, accuracy in subset classification is in the range of 90% both in trial and validation cohorts. Importantly, these mutations can also be detected in the blood, where tumor cells may be found in the great majority of patients, indicating that it is possible to classify disease before splenectomy, using a “liquid biopsy” approach.
Moving from genes to patterns of expression and to morphology, the authors show that the DMT cluster is characterized by an atypical morphology, whereas cases belonging to the other 3 categories display a more typical morphology. Subsequent analysis of expression of 1402 “microenvironmental” genes by RNA sequencing added an additional layer to this novel molecular stratification, by dividing SMZL in immune suppressive and immune silent subsets. The first subset is characterized by expression of genes typical of T cells, type 2 macrophages, and coding for exhaustion markers. These findings are in line with the concept that infiltrating immune cells cannot react against the tumor. Conversely, the immune silent subset essentially lacks expression of microenvironment-related genes, with an overt dominance of pathways belonging to tumor B cells, underlining that these tumors are not invaded by T cells. Interestingly, these expression profiles are equally distributed among genetic profiles, raising the question of what controls microenvironment organization. Mouse models recapitulating the different genetic lesions will likely be instrumental in providing additional information on how tumor cells shape the microenvironment.6,7 In addition, a possible role of infectious agents, for SMZL typically hepatitis C virus, needs to be addressed to determine whether it can induce a T-cell-rich and exhausted, or a T-cell–depleted environment.
In summary, this comprehensive study reclassified SMZL, complementing classical diagnostic tests, such as immunohistochemistry and flow cytometry, with mutational analyses and gene expression profiles. Survival data show that this classification is effective in identifying disease subgroups with differing overall survival. Even if in the case of an indolent disease such as SMZL the clinical impact of genomic and transcriptomic (re)classification deriving from of this observation may be limited, it will certainly be a founding element for the design of novel therapeutic strategies, as is already happening in other more aggressive lymphomas.8
There are 2 main features of this work that make it applicable for clinical care. The first is the definition of a minimal set of 14 genes that created an accurate mutational classification of SMZL into the 4 subentities described previously. This panel is easily adaptable to routine diagnosis of pathology laboratories. The second feature is that, in principle, the panel could be run using peripheral blood samples. If confirmed by independent studies, these findings imply that mutational classification could be performed independent of splenectomy. Treatment of SMZL is currently shifting from splenectomy to more conservative approaches that target the B cell: future studies will tell whether different mutational profiles will respond to different targeted drugs.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal