

Key Points
Fixed-duration ibrutinib plus venetoclax achieved deep, durable responses, clinically meaningful PFS, and treatment-free remissions.
First-line ibrutinib plus venetoclax represents the first all-oral, once-daily, chemotherapy-free, fixed-duration regimen for CLL.

Abstract
CAPTIVATE (NCT02910583) is an international phase 2 study in patients aged ≤70 years with previously untreated chronic lymphocytic leukemia (CLL). Results from the cohort investigating fixed-duration (FD) treatment with ibrutinib plus venetoclax are reported. Patients received 3 cycles of ibrutinib lead-in then 12 cycles of ibrutinib plus venetoclax (oral ibrutinib [420 mg/d]; oral venetoclax [5-week ramp-up to 400 mg/d]). The primary endpoint was complete response (CR) rate. Hypothesis testing was performed for patients without del(17p) with prespecified analyses in all treated patients. Secondary endpoints included undetectable minimal residual disease (uMRD) rates, progression-free survival (PFS), overall survival (OS), and safety. Of the 159 patients enrolled and treated, 136 were without del(17p). The median time on study was 27.9 months, and 92% of patients completed all planned treatment. The primary endpoint was met, with a CR rate of 56% (95% confidence interval [CI], 48-64) in patients without del(17p), significantly higher than the prespecified 37% minimum rate (P < .0001). In the all-treated population, CR rate was 55% (95% CI, 48-63); best uMRD rates were 77% (peripheral blood [PB]) and 60% (bone marrow [BM]); 24-month PFS and OS rates were 95% and 98%, respectively. At baseline, 21% of patients were in the high tumor burden category for tumor lysis syndrome (TLS) risk; after ibrutinib lead-in, only 1% remained in this category. The most common grade ≥3 adverse events (AEs) were neutropenia (33%) and hypertension (6%). First-line ibrutinib plus venetoclax represents the first all-oral, once-daily, chemotherapy-free FD regimen for patients with CLL. FD ibrutinib plus venetoclax achieved deep, durable responses and promising PFS, including in patients with high-risk features.
Introduction
Ibrutinib, a once-daily oral Bruton’s tyrosine kinase (BTK) inhibitor, is the only targeted therapy to demonstrate improved progression-free survival (PFS) in 4 randomized phase 3 studies,1-4 with improved overall survival (OS) in 2 of these studies1,2 vs standard chemotherapy/ chemoimmunotherapy regimens in previously untreated chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL). Venetoclax, an oral inhibitor of the antiapoptotic protein BCL-2, is approved for the treatment of CLL and SLL as a single agent or combined with rituximab or obinutuzumab.5 Single-agent venetoclax produces deep responses with undetectable minimal residual disease (uMRD) status in bone marrow (BM) in 16% of treated patients with relapsed and/or refractory CLL.6-8 While continuous ibrutinib is an established standard of care in CLL that affords survival benefit, there is increasing desire for convenient, all-oral, time-limited treatment options that can be delivered in the outpatient setting.
Ibrutinib and venetoclax, through distinct and complementary modes of action, preferentially target distinct cell compartments and CLL subpopulations to eliminate both dividing and resting CLL cells.9,10 Ibrutinib mobilizes CLL cells out of lymph nodes and other lymphoid niches and into peripheral blood (PB), where they are more susceptible to venetoclax-induced apoptosis.9,11-14 Inhibition of BTK by ibrutinib also enhances the dependence of CLL cells on BCL-2, thereby increasing sensitivity to venetoclax and accelerating apoptosis.9,13,15 Combined ibrutinib plus venetoclax demonstrated synergistic antitumor activity in preclinical CLL models, with greater cytotoxicity observed with the combination than with either agent alone.9,12,15 Additionally, recent clinical studies with ibrutinib plus venetoclax demonstrated high uMRD rates in both PB and BM in patients with CLL or SLL.16-22
CAPTIVATE is a multicenter phase 2 study investigating combined ibrutinib plus venetoclax in first-line treatment of CLL and SLL in 2 separate cohorts: MRD-guided randomized treatment discontinuation (MRD) and fixed-duration (FD). In the MRD cohort, uMRD was achieved in 75% of patients in PB and 68% in BM after 3 cycles of ibrutinib followed by 12 cycles of ibrutinib plus venetoclax, and 30-month PFS rates were consistently ≥95% across subsequent MRD-guided randomized treatments.17 Here, we report primary analysis results from the CAPTIVATE FD cohort evaluating FD ibrutinib plus venetoclax for the first-line treatment of CLL or SLL.
Methods
Study design and participants
CAPTIVATE is a multicenter, international, phase 2 study. The FD cohort enrolled sequentially after the MRD cohort and is an open-label, single-arm cohort to evaluate the depth of response with FD ibrutinib plus venetoclax. Eligible patients were aged ≥18 and ≤70 years with previously untreated CLL or SLL requiring treatment by International Workshop on Chronic Lymphocytic Leukemia (iwCLL) 2008 criteria23 and had measurable nodal disease by computed tomography (CT); Eastern Cooperative Oncology Group performance status of 0 to 2; and adequate hepatic, renal, and hematologic function. Patients with known allergy to xanthine oxidase inhibitors and/or rasburicase were excluded. The study was conducted in accordance with the International Conference on Harmonization guidelines for Good Clinical Practice and principles of the Declaration of Helsinki. The protocol was approved by institutional review boards or independent ethics committees of all participating institutions. All patients provided written informed consent. This study is registered with ClinicalTrials.gov (NCT02910583).
Treatment
Patients in the FD cohort received all-oral FD treatment with 3 cycles of single-agent ibrutinib (420 mg once daily) followed by 12 cycles of combined ibrutinib plus venetoclax (target dose 400 mg once daily after standard 5-week ramp-up, with tumor lysis syndrome [TLS] prophylaxis and monitoring per US prescribing information).5 Treatment was administered in 28-day cycles. After completion of the FD regimen, patients who subsequently had confirmed progressive disease (PD) by iwCLL criteria could be retreated with single-agent ibrutinib until PD or unacceptable toxicity. For patients who had PD >2 years after completion of the FD regimen, retreatment with the FD ibrutinib plus venetoclax regimen could be considered.
Outcomes and assessments
The primary endpoint was complete response (CR) rate (proportion of patients with CR or CR with incomplete BM recovery [CRi]) for patients without del(17p) assessed by investigators by 2008 iwCLL criteria,23,24 with prespecified supporting analysis in all treated patients. Secondary endpoints were duration of response (time from initial documentation of response until PD or death from any cause), uMRD rates in PB and BM (proportion of patients with <1 CLL cell per 10 000 leukocytes), overall response rate (proportion of patients with partial response [PR] or better) by iwCLL criteria, reduction in tumor burden category for TLS prophylaxis (proportion of patients in high tumor burden category after 3-cycle ibrutinib lead-in vs at baseline), PFS, OS, and safety and tolerability in all treated patients. Sensitivity analyses were performed of CR rates and PFS per independent review committee (IRC) assessment.
Clinical response and progression were assessed by physical examination, hematologic parameters, and radiographic evaluation (CT or magnetic resonance imaging [MRI]); BM biopsy was performed at cycle 10, at cycle 19, and/or if there was suspected CR on other assessments. Confirmation of response by CT or MRI was required per protocol. Overall response assessments were performed at the end of cycle 3, on day 1 of cycles 7, 10, 13, 19, 25, 28, and 31, and every 6 months thereafter. MRD status was evaluated by central laboratory per European Research Initiative on CLL recommendations25 using 8-color flow cytometry in PB samples obtained at baseline, on day 1 of cycles 7, 10, and 13, at the end of treatment (EOT) (30 days after discontinuation), on day 1 of cycles 19, 25, 28, and 31, and annually thereafter; and in BM samples obtained on day 1 of cycles 10 and 19. Tumor burden categories for TLS prophylaxis were assessed at baseline and before initiation of venetoclax in cycle 4 using criteria described in venetoclax US prescribing information.5
Safety assessments included adverse events (AEs), laboratory evaluations, and physical examinations. AEs were monitored throughout treatment until 30 days after the last dose of study treatment. Hematological AEs were graded using iwCLL criteria.23 Nonhematological AEs were graded using National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03.26 TLS was assessed using Howard criteria.27
Cytogenetics were evaluated by a central laboratory or by a local laboratory, with central laboratory results used where available. Karyotype, immunoglobulin heavy chain variable region (IGHV) gene mutational status, and mutational status of other relevant genes, including TP53, were assessed by a central laboratory (supplemental Methods, available on the Blood Web site).
Statistical analysis
The sample size was calculated based on the primary endpoint of CR rate in patients without del(17p). Assuming the CR rate for ibrutinib plus venetoclax is 50%, 125 patients without del(17p), irrespective of TP53 mutation status, would provide 83% power to exclude a minimum CR rate of 37% at a 1-sided α of .025. A CR rate of 50% would represent a significant improvement over FD treatment with bendamustine plus rituximab (BR) (31%) and meaningful improvement over fludarabine, cyclophosphamide, and rituximab (FCR) (40%) in the CLL10 study, which excluded patients with del(17p).28
Efficacy and safety were evaluated in all patients who received ≥1 dose of study treatment with hypothesis testing performed for patients without del(17p). Subgroup analyses by prognostic factors, including del(17p) and/or mutated TP53, were prespecified in the statistical analysis plan. Time-to-event endpoints were estimated using the Kaplan-Meier method; other efficacy endpoints and AEs were summarized descriptively. The 95% CIs for response rates were estimated based on the normal approximation to the binomial distribution.
Results
Patients
A total of 159 patients were enrolled, including 136 patients without del(17p). Median age was 60 years (range, 33-71) (Table 1). A high proportion of patients had 1 or more high-risk disease features, including del(17p) (n = 20; 13%), del(17p) and/or mutated TP53 (n = 27; 17%), complex karyotype (n = 31; 19%), unmutated IGHV (n = 89; 56%), or del(11q) without del(17p) (n = 28; 18%); 48 patients (30%) had bulky disease (Table 1).
Patient demographics and disease characteristics at baseline
| Characteristic | All treated patients (n = 159), n (%) |
|---|---|
| Age | |
| Median, y (range) | 60 (33-71) |
| ≥65 y | 45 (28) |
| Male | 106 (67) |
| ECOG PS | |
| 0 | 110 (69) |
| 1 | 49 (31) |
| Histology | |
| CLL | 146 (92) |
| SLL | 13 (8) |
| Rai stage | |
| 0/I/II | 113 (71) |
| III/IV | 44 (28) |
| Missing | 2 (1) |
| Bulky disease (cm) | |
| ≥5 | 48 (30) |
| ≥10 | 5 (3) |
| Cytopenia at baseline | |
| Any cytopenia | 54 (34) |
| Hemoglobin ≤11 g/dL | 37 (23) |
| Platelet count ≤100 × 109/L | 21 (13) |
| ANC ≤1.5 × 109/L | 13 (8) |
| Hierarchical cytogenetics (FISH) classification* | |
| Del(17p) | 20 (13) |
| Del(11q) | 28 (18) |
| Trisomy 12 | 23 (14) |
| Normal | 33 (21) |
| Del(13q) | 54 (34) |
| Unknown | 1 (1) |
| Mutated TP53 | |
| Yes | 16 (10) |
| No | 142 (89) |
| Unknown | 1 (1) |
| Del(17p) or mutated TP53 | |
| Yes | 27 (17) |
| No | 129 (81) |
| Unknown | 3 (2) |
| IGHV gene mutation status | |
| Unmutated | 89 (56) |
| Mutated | 66 (42) |
| Unknown | 4 (3) |
| Complex karyotype† | |
| Yes | 31 (19) |
| No | 102 (64) |
| Unknown | 26 (16) |
| Characteristic | All treated patients (n = 159), n (%) |
|---|---|
| Age | |
| Median, y (range) | 60 (33-71) |
| ≥65 y | 45 (28) |
| Male | 106 (67) |
| ECOG PS | |
| 0 | 110 (69) |
| 1 | 49 (31) |
| Histology | |
| CLL | 146 (92) |
| SLL | 13 (8) |
| Rai stage | |
| 0/I/II | 113 (71) |
| III/IV | 44 (28) |
| Missing | 2 (1) |
| Bulky disease (cm) | |
| ≥5 | 48 (30) |
| ≥10 | 5 (3) |
| Cytopenia at baseline | |
| Any cytopenia | 54 (34) |
| Hemoglobin ≤11 g/dL | 37 (23) |
| Platelet count ≤100 × 109/L | 21 (13) |
| ANC ≤1.5 × 109/L | 13 (8) |
| Hierarchical cytogenetics (FISH) classification* | |
| Del(17p) | 20 (13) |
| Del(11q) | 28 (18) |
| Trisomy 12 | 23 (14) |
| Normal | 33 (21) |
| Del(13q) | 54 (34) |
| Unknown | 1 (1) |
| Mutated TP53 | |
| Yes | 16 (10) |
| No | 142 (89) |
| Unknown | 1 (1) |
| Del(17p) or mutated TP53 | |
| Yes | 27 (17) |
| No | 129 (81) |
| Unknown | 3 (2) |
| IGHV gene mutation status | |
| Unmutated | 89 (56) |
| Mutated | 66 (42) |
| Unknown | 4 (3) |
| Complex karyotype† | |
| Yes | 31 (19) |
| No | 102 (64) |
| Unknown | 26 (16) |
ANC, absolute neutrophil count; ECOG PS, Eastern Cooperative Oncology Group performance status; FISH, fluorescence in situ hybridization; MRD, minimal residual disease.
Per Döhner hierarchy.
Defined as ≥3 abnormalities by CpG-stimulated cytogenetics.
The median time on study was 27.9 months (range, 0.8-33.2); 153 of 159 patients completed 3 cycles of ibrutinib lead-in and started treatment with ibrutinib plus venetoclax (Figure 1). Overall, 92% (147/159) of patients completed the planned 12 cycles of combined ibrutinib plus venetoclax.
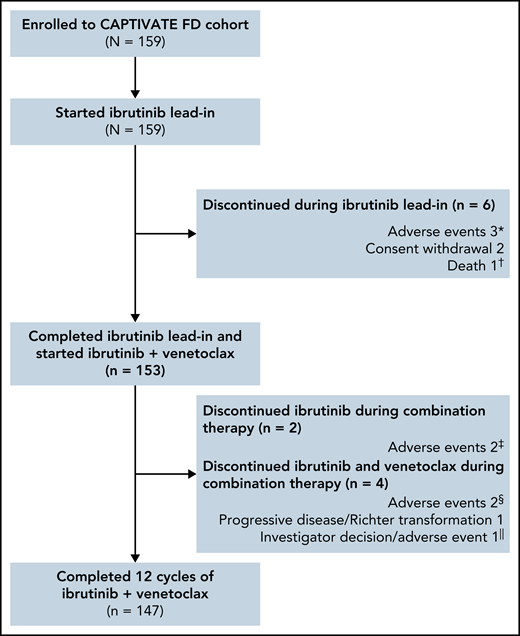
Patient flow and disposition. *Hepatitis, erythematous rash, and cerebral hemorrhage in 1 patient each. †Sudden death on study day 23 in a 54-year-old male with a medical history of hypertension, hyperlipidemia, smoking, fatigue, anxiety, depression, insomnia, and gastroesophageal reflux disease. ‡Hemorrhagic cerebral infarction and sinus arrest in 1 patient each. §Pneumonia and neutropenia + pharyngitis in 1 patient each. ǁPatient discontinued venetoclax due to an AE (nausea + vomiting) and subsequently discontinued ibrutinib due to investigator decision.
Patient flow and disposition. *Hepatitis, erythematous rash, and cerebral hemorrhage in 1 patient each. †Sudden death on study day 23 in a 54-year-old male with a medical history of hypertension, hyperlipidemia, smoking, fatigue, anxiety, depression, insomnia, and gastroesophageal reflux disease. ‡Hemorrhagic cerebral infarction and sinus arrest in 1 patient each. §Pneumonia and neutropenia + pharyngitis in 1 patient each. ǁPatient discontinued venetoclax due to an AE (nausea + vomiting) and subsequently discontinued ibrutinib due to investigator decision.
Efficacy
The primary endpoint was met with a CR rate of 56% (95% CI, 48-64) by investigator assessment in patients without del(17p) (Figure 2A); this rate significantly exceeds the minimum CR rate of 37% (1-sided P < .0001) and 40% rate of the historical FCR comparator.28 CR rates were 55% (95% CI, 48-63) in the all-treated population and 56% (95% CI, 37-74) in patients with del(17p) and/or mutated TP53 (Figure 2A). CR rates by IRC assessment were 60% (95% CI, 52-67) in the all-treated population, 61% (95% CI, 53-69) in patients without del(17p), and 56% (95% CI, 37-74) in patients with del(17p) and/or mutated TP53. CR rates by investigator assessment were high and generally consistent across subgroups defined by baseline characteristics (Figure 2B); the CR rate was higher in patients without vs with bulky disease ≥5 cm (66% vs 31%) and in those with unmutated vs mutated IGHV (62% vs 47%). CR rates at 3 months after EOT are shown in supplemental Table 1. Consistent with CR rates, best overall response rates by investigator assessment were 96% (95% CI, 93-99) in the all-treated population, 96% (95% CI, 92-99) in patients without del(17p), and 96% (95% CI, 89-100) in patients with del(17p) and/or mutated TP53 (Figure 2A). Among patients in the all-treated population with the best response of PR or nodular PR (nPR), 80% (52/65) had lymph node diameter >1.5 cm. Among 88 patients in the all-treated population who achieved CR by investigator assessment, 78/79 (99%) with ≥12 cycles of follow-up had CR lasting ≥12 cycles; 1 patient died of cardiac arrest 7 months after initial CR and ∼1.5 months after completion of treatment.
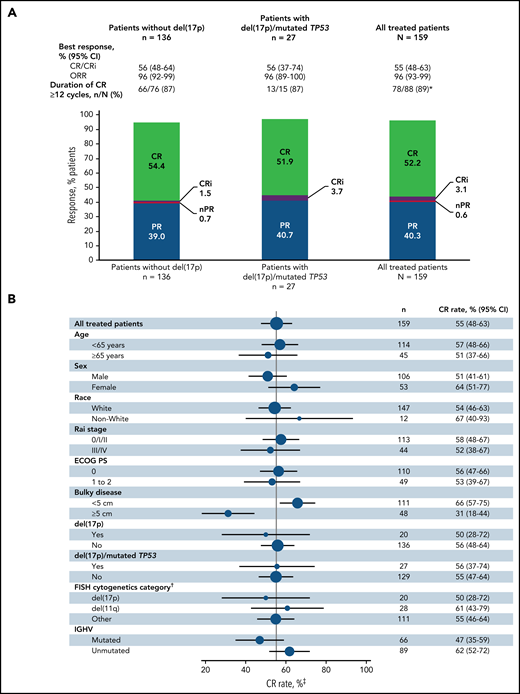
Best overall response. (A) Best overall response rates as assessed by the investigator in the all-treated population and patients without del(17p) or with del(17p)/mutated TP53. (B) Forest plots of investigator-assessed CR rates across patient subgroups by baseline characteristics in the all-treated population. DOCR, duration of CR; ECOG PS, Eastern Cooperative Oncology Group performance status; FISH, fluorescence in situ hybridization; ORR, overall response rate; nPR, nodular partial response; PR, partial response. *After achieving CR, 9 patients with <1 year of follow-up were not evaluable; 1 patient died of cardiac arrest 7 months after initial CR and 47 days after completion of therapy. †Per Döhner hierarchy. ‡Proportion of patients with CR or CRi.
Best overall response. (A) Best overall response rates as assessed by the investigator in the all-treated population and patients without del(17p) or with del(17p)/mutated TP53. (B) Forest plots of investigator-assessed CR rates across patient subgroups by baseline characteristics in the all-treated population. DOCR, duration of CR; ECOG PS, Eastern Cooperative Oncology Group performance status; FISH, fluorescence in situ hybridization; ORR, overall response rate; nPR, nodular partial response; PR, partial response. *After achieving CR, 9 patients with <1 year of follow-up were not evaluable; 1 patient died of cardiac arrest 7 months after initial CR and 47 days after completion of therapy. †Per Döhner hierarchy. ‡Proportion of patients with CR or CRi.
Best uMRD rates were 77% (95% CI, 70-83) in PB and 60% (95% CI, 52-67) in BM in the all-treated population (Figure 3A). In patients without del(17p), uMRD rates were 76% (95% CI, 69-84) in PB and 62% (95% CI, 54-70) in BM. uMRD rates for patients with del(17p) and/or mutated TP53 were 81% (95% CI, 67-96) in PB and 41% (95% CI, 22-59) in BM. In the all-treated population, uMRD rates in BM were consistent across patient subgroups based on baseline characteristics (Figure 3B); in contrast to CR rates, uMRD rates were similar in PB (both 77%) and BM (59% vs 63%) in patients without vs with bulky disease. uMRD rates were higher in patients with unmutated vs mutated IGHV in PB (84% vs 67%) and BM (64% vs 53%). In patients with best response of CR (n = 88), uMRD rates were 90% in PB and 72% in BM; in patients with PR or nPR (n = 65), corresponding rates were 63% and 49%, respectively. Overall, 40% (63/159) of patients had CR and uMRD in BM. uMRD rates at 3 months after EOT in the all-treated population and in patients with CR are shown in supplemental Table 1.
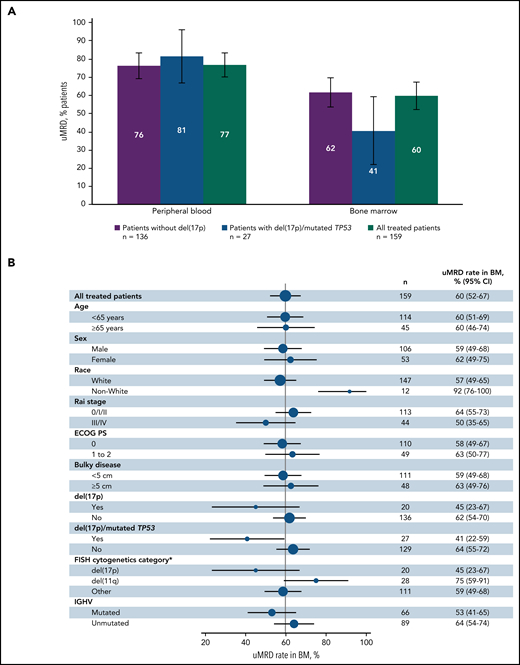
Best MRD response. (A) Rates of undetectable MRD in PB and BM in patients without del(17p) or with del(17p) and/or mutated TP53. (B) Forest plot of undetectable MRD in BM across patient subgroups by baseline characteristics in the all-treated population. Error bars represent 95% CIs. *Per Döhner hierarchy.
Best MRD response. (A) Rates of undetectable MRD in PB and BM in patients without del(17p) or with del(17p) and/or mutated TP53. (B) Forest plot of undetectable MRD in BM across patient subgroups by baseline characteristics in the all-treated population. Error bars represent 95% CIs. *Per Döhner hierarchy.
With median follow-up of 27.9 months, estimated 24-month PFS rates by investigator assessment were 95% (95% CI, 90-97) in the all-treated population, 96% (95% CI, 91-98) in patients without del(17p) (Figure 4A), and 84% (95% CI, 63-94) in patients with del(17p)/mutated TP53. PD events are summarized in supplemental Table 2. Estimated 24-month PFS rates were 93% (95% CI, 85-97) for patients with unmutated IGHV vs 97% (95% CI, 88-99) for patients with mutated IGHV. PFS by IRC assessment was similar to that assessed by investigators (supplemental Figure 1). PFS rates by investigator assessment remained high with additional follow-up (supplemental Figure 2). In the all-treated population, 24-month PFS rates were high (97%-100%) regardless of clinical response (CR including CRi or PR including nPR) and MRD status in BM (undetectable or detectable) at 3 months after EOT (supplemental Table 3). Estimated 24-month OS rates were 98% (95% CI, 94-99) in the all-treated population, 98% (95% CI, 93-99) in patients without del(17p) (Figure 4B), and 96% (95% CI, 76-99) in patients with del(17p)/mutated TP53.
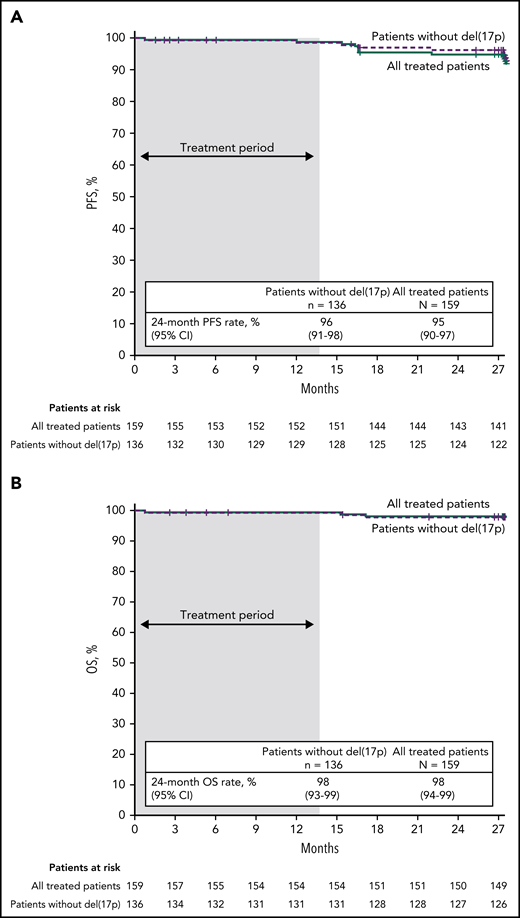
PFS and OS. Kaplan-Meier curves of (A) PFS as assessed by investigators and (B) OS in the all-treated population and patients without del(17p). Tick marks indicate patients with censored data. Shading indicates time on treatment.
PFS and OS. Kaplan-Meier curves of (A) PFS as assessed by investigators and (B) OS in the all-treated population and patients without del(17p). Tick marks indicate patients with censored data. Shading indicates time on treatment.
Of the patients with PD after FD treatment with ibrutinib plus venetoclax, next-generation sequencing found no evidence of BTK, PLCg2, or BCL-2 mutations associated with resistance to ibrutinib or venetoclax in 13/13 patients with available data (supplemental Table 2). Three variants (2 in PLCg2 and 1 in BCL-2) were present at similar levels at baseline and progression; 2 of them were common single-nucleotide polymorphisms, and none of the 3 variants have been previously associated with PD on ibrutinib or venetoclax treatment.
As of 4 August 2021, after experiencing PD, 9 patients have been retreated with single-agent ibrutinib. Of the patients retreated with single-agent ibrutinib, 7/9 patients have the best response of PR, and 2 patients are pending response evaluation. One additional patient, who progressed after completing FD treatment with ibrutinib plus venetoclax, received subsequent therapy with acalabrutinib, with response unknown.
Tumor debulking
At baseline risk assessment for TLS, 21% (34/159) of patients were categorized as having a high tumor burden. After 3 cycles of ibrutinib lead-in, 1% (1/159) of patients were in the high tumor burden category for TLS risk (Figure 5A); effective debulking shifted 94% from high to medium or low tumor burden categories, and no patients shifted from medium or low to high. The proportion of patients indicated for hospitalization for TLS monitoring and prophylaxis per venetoclax prescribing information decreased from 40% (63/159) at baseline to 18% (28/159) after ibrutinib lead-in (Figure 5B).
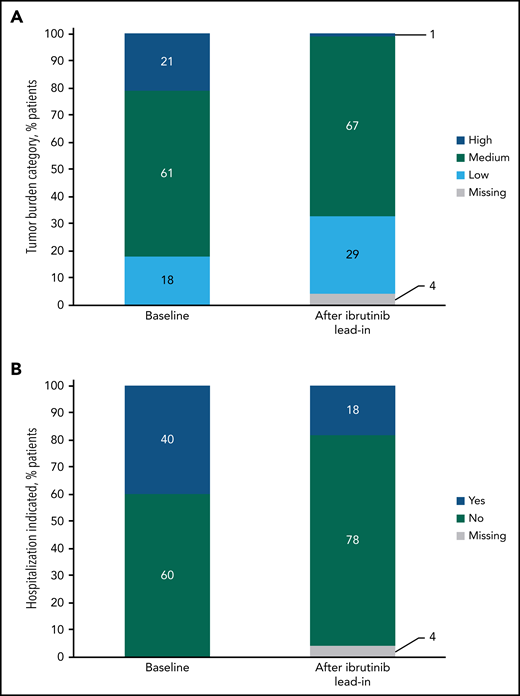
Impact of single-agent ibrutinib lead-in on tumor burden for TLS prophylaxis. (A) Tumor burden categories for TLS prophylaxis at baseline and after ibrutinib lead-in in the all-treated population. (B) Indication for hospitalization (*) for TLS monitoring and prophylaxis at baseline and after ibrutinib lead-in in the all-treated population. *Defined as patients in high tumor burden category for TLS prophylaxis or patients in medium tumor burden category with creatinine clearance (CrCL) <80 mL/min. Of 28 patients who met the criteria for indication for hospitalization after lead-in, 27 had medium tumor burden and a CrCL of <80 mL/min, and 1 had high tumor burden.
Impact of single-agent ibrutinib lead-in on tumor burden for TLS prophylaxis. (A) Tumor burden categories for TLS prophylaxis at baseline and after ibrutinib lead-in in the all-treated population. (B) Indication for hospitalization (*) for TLS monitoring and prophylaxis at baseline and after ibrutinib lead-in in the all-treated population. *Defined as patients in high tumor burden category for TLS prophylaxis or patients in medium tumor burden category with creatinine clearance (CrCL) <80 mL/min. Of 28 patients who met the criteria for indication for hospitalization after lead-in, 27 had medium tumor burden and a CrCL of <80 mL/min, and 1 had high tumor burden.
Safety
The median duration of treatment was 13.8 months (range, 0.5-24.9), corresponding to 15 28-day treatment cycles. The most common treatment-emergent AEs were diarrhea (62%), nausea (43%), neutropenia (42%), and arthralgia (33%) (Table 2); AEs were primarily grade 1 or 2 in severity. The most common grade 3/4 AEs were neutropenia (33%), hypertension (6%), and neutrophil count decreased (5%). One fatal AE (sudden death) occurred during ibrutinib lead-in. Serious AEs occurred in 36 patients (23%) (supplemental Table 4).
Treatment-emergent AEs
| AEs | All treated patients (n = 159), n (%) | |
|---|---|---|
| Any grade | Grade 3/4 | |
| Most common AEs* | ||
| Diarrhea | 99 (62) | 5 (3) |
| Nausea | 68 (43) | 2 (1) |
| Neutropenia | 66 (42) | 52 (33) |
| Arthralgia | 53 (33) | 2 (1) |
| Hypertension | 25 (16) | 9 (6) |
| Neutrophil count decreased | 16 (10) | 8 (5) |
| Other AEs of clinical interest | ||
| Atrial fibrillation | 7 (4) | 2 (1) |
| Major hemorrhage† | 3 (2) | 2 (1) |
| Laboratory safety parameters | ||
| Hematology | ||
| Neutrophils decreased | 115 (72) | 60 (38) |
| Platelets decreased | 94 (59) | 20 (13) |
| Hemoglobin decreased | 31 (19) | 0 |
| Chemistry | ||
| Corrected calcium decreased | 61 (38) | 1 (1) |
| Potassium increased | 39 (25) | 4 (3) |
| Uric acid increased | 34 (21) | 34 (21) |
| Creatinine increased | 27 (17) | 0 |
| AEs | All treated patients (n = 159), n (%) | |
|---|---|---|
| Any grade | Grade 3/4 | |
| Most common AEs* | ||
| Diarrhea | 99 (62) | 5 (3) |
| Nausea | 68 (43) | 2 (1) |
| Neutropenia | 66 (42) | 52 (33) |
| Arthralgia | 53 (33) | 2 (1) |
| Hypertension | 25 (16) | 9 (6) |
| Neutrophil count decreased | 16 (10) | 8 (5) |
| Other AEs of clinical interest | ||
| Atrial fibrillation | 7 (4) | 2 (1) |
| Major hemorrhage† | 3 (2) | 2 (1) |
| Laboratory safety parameters | ||
| Hematology | ||
| Neutrophils decreased | 115 (72) | 60 (38) |
| Platelets decreased | 94 (59) | 20 (13) |
| Hemoglobin decreased | 31 (19) | 0 |
| Chemistry | ||
| Corrected calcium decreased | 61 (38) | 1 (1) |
| Potassium increased | 39 (25) | 4 (3) |
| Uric acid increased | 34 (21) | 34 (21) |
| Creatinine increased | 27 (17) | 0 |
AEs of any grade occurring in ≥30% of patients or grade 3/4 occurring in ≥5% of patients.
Major hemorrhage was identified using the Standardized MedDRA Query for Hemorrhage, excluding laboratory terms.
Among other AEs of clinical interest, atrial fibrillation of any grade occurred in 7 patients (4%) and was grade ≥3 in 2 patients (1%) (Table 2). Of these patients, 3 had resolution and 4 had ongoing grade 1 or 2 events at the time of analysis. Major hemorrhage occurred in 3 patients (2%), with cerebral hemorrhage, hemorrhagic cerebral infarction, and retinal hemorrhage in 1 patient each. Bleeding events of any grade occurred in 37/61 patients (61%) receiving concomitant anticoagulants/antiplatelets and 58/98 patients (59%) not receiving anticoagulants/antiplatelets and were grade 3/4 in 0/58 and 2/98 patients (2%), respectively. Infections of any grade occurred in 106 patients (67%), with grade ≥3 infections in 13 patients (8%). Febrile neutropenia (grade 4) occurred in 1 patient. No clinical TLS was reported.
Frequencies of hematologic laboratory abnormalities were consistent with the AE profile of the ibrutinib plus venetoclax combination, and frequencies of abnormalities in individual laboratory parameters monitored for TLS were modest (Table 2); no laboratory TLS events were identified by Howard criteria. Overall, per protocol and investigator discretion following local standards of care, 26 patients (16%) were hospitalized for TLS monitoring and prophylaxis. Rasburicase was used for TLS prophylaxis in 8 patients (5%) (supplemental Table 5).
AEs led to dose reductions of ibrutinib only in 9 patients (6%), venetoclax only in 18 patients (11%), and both ibrutinib and venetoclax in 6 patients (4%) (supplemental Table 4). Of 33 patients with AEs leading to dose reduction, 88% had resolution at the time of analysis. AEs led to discontinuation of ibrutinib only in 5 patients (3%), venetoclax only in 1 patient (1%), and both ibrutinib and venetoclax in 2 patients (1%) (supplemental Table 4). With 92% of patients able to complete the full FD regimen, ibrutinib plus venetoclax appears tolerable regardless of common concomitant medications, notably including anticoagulants (11%), antiplatelet agents (31%), antihypertensives (36%), and acid-reducing agents (44%), such as proton pump inhibitors (30%) (supplemental Table 5).
Discussion
Results from the FD cohort of the CAPTIVATE study demonstrate that first-line treatment with an FD regimen of 3 cycles of single-agent ibrutinib followed by 12 cycles of ibrutinib plus venetoclax, the first all-oral, FD combination regimen for patients with CLL, provides a high CR rate, including in those with high-risk genomic features. The primary endpoint was met, with a CR rate of 56% (95% CI, 48-64) by investigator assessment in patients without del(17p), exceeding historical rates with the 2 most efficacious chemoimmunotherapy comparators of BR (31%) and FCR (40%) in a similar patient population without del(17p) in the CLL10 study, noting that CLL10 included patients aged >70 years.28 CR rate was consistent with those previously reported with first-line ibrutinib plus venetoclax and is similar to that observed with venetoclax plus obinutuzumab (50%), albeit in a different patient population.16,18,29 Importantly, CR was maintained for ≥12 cycles in 78/79 patients (99%) with sufficient follow-up.
In addition to high CR rates, ibrutinib plus venetoclax achieved high rates of uMRD in both PB and BM at the standard cutoff of <1 CLL cell per 10 000 leukocytes. Results to date with FD regimens suggest that achievement of uMRD may be a stronger predictor of PFS than the achievement of CR.30-33 The high uMRD rates in the FD cohort are consistent with those previously reported for the MRD cohort with 12 cycles of combined ibrutinib plus venetoclax (68% in BM and 75% in PB),16 as well as those reported in a phase 2 study of first-line ibrutinib plus venetoclax (56% in BM after 12 cycles of the combination).18 uMRD rates also compare favorably with those obtained with FCR, the most efficacious FD chemoimmunotherapy regimen for first-line CLL (43% in BM and 59% to 63% in PB),2,30,31,33 and are similar to those observed with venetoclax plus obinutuzumab (57% in BM and 76% in PB 3 months after EOT).29 Analysis of MRD at lower limits of detection remains to be done for CAPTIVATE. CR and uMRD rates were high across patient subgroups, including those with high-risk disease features, such as del(17p) and/or mutated TP53, del(11q), and unmutated IGHV gene. Interestingly, the higher CR and uMRD rates observed in high-risk unmutated IGHV patients vs those with mutated IGHV may reflect the impact of ibrutinib’s inhibition of the BCR pathway, which is relevant for these CLL clones.
With a median follow-up of 28 months in the CLL14 study, the 24-month PFS rate was 88% with FD venetoclax plus obinutuzumab in an older/less fit patient population.29 In the CAPTIVATE FD cohort, the 24-month PFS rate of 95%, together with results from the CAPTIVATE MRD cohort demonstrating 30-month PFS rates of ≥95% in placebo-randomized patients with confirmed uMRD given MRD-guided treatment,17 supports the potential for durable, treatment-free remissions with FD ibrutinib plus venetoclax in younger patients who may prefer time-limited treatment. Patients in the CAPTIVATE study represent a relatively young, fit population (aged ≤70 years with creatinine clearance (CrCL) ≥60 mL/min) typical of patients who may be considered eligible for treatment with FCR. Consequently, CAPTIVATE results may not be generalizable to the overall population of patients with CLL who tend to be older and have more comorbidities. However, the randomized phase 3 GLOW study (NCT03462719) evaluated this regimen in a complementary population of unfit/elderly patients (aged ≥65 years or 18-64 years with cumulative illness rating scale score >6 or CrCL <70 mL/min) with previously untreated CLL/SLL. Primary analysis results from the GLOW study demonstrated significantly improved PFS and greater depth of remission (CR rates and uMRD rates) with ibrutinib plus venetoclax compared with chlorambucil plus obinutuzumab.34
Tumor debulking with the 3 cycles of ibrutinib lead-in reduced tumor burden category for TLS monitoring and prophylaxis for 94% of patients with high tumor burden at baseline. Consequently, hospitalization with venetoclax initiation was no longer indicated in more than half of patients with such indication at baseline. This is the first regimen to demonstrate this benefit to patients when combined with venetoclax. Additionally, laboratory parameters monitored for TLS were less frequently abnormal during the overall study period than reported with venetoclax plus obinutuzumab in CLL14.29 With a reduced need for intensive monitoring and hospitalization and no risk of infusion-related reactions, the all-oral, FD regimen of ibrutinib plus venetoclax offers improved patient convenience and ease of administration.
Ibrutinib plus venetoclax had a favorable safety profile, with 92% of patients able to complete all planned treatment with the FD regimen. The safety profile of ibrutinib plus venetoclax in the FD cohort was similar to that observed in the MRD cohort17 and consistent with known safety profiles for each agent alone; no new safety signals were observed. Incidences of diarrhea and neutropenia were somewhat higher than expected with ibrutinib alone. However, most diarrhea events were grade 1/2 in severity, with grade ≥3 diarrhea in only 3% of patients; the frequency of grade ≥3 neutropenia (33%) was generally similar to that observed with single-agent venetoclax (37%).35 All patients who had dose reductions for diarrhea or neutropenia had resolution of the AE without discontinuation. The combination of ibrutinib plus venetoclax was well tolerated in this population of young, fit patients, evidenced by the low rate of discontinuations due to AEs (5%). Similarly, in the unfit or elderly population of the randomized, phase 3 GLOW study, 77% of patients were able to complete all planned treatment with ibrutinib plus venetoclax.34 The most common grade ≥3 AEs were neutropenia or neutrophil count decreased (35%), diarrhea (10%), and hypertension (8%) with ibrutinib plus venetoclax, and neutropenia or neutrophil count decreased (50%), thrombocytopenia (20%), and pneumonia and TLS (6% each) with chlorambucil plus obinutuzumab; grade 5 AEs occurred in 7/106 patients on ibrutinib plus venetoclax and 2/105 patients on chlorambucil plus obinutuzumab.34
Importantly, promising clinical responses have been observed in early data of single-agent ibrutinib retreatment in patients with PD after the FD regimen. Additionally, DNA sequencing of CLL cells from relapsing patients found no resistance-associated mutations in BTK, PLCg2, or BCL-2. Together these results suggest that retreatment after FD ibrutinib plus venetoclax may be a feasible strategy to provide extended clinical benefit.
In summary, first-line ibrutinib plus venetoclax is an all-oral, once-daily, chemotherapy-free FD regimen that drives deep, durable responses and can be delivered in the outpatient setting for most young, fit patients with CLL or SLL. Overall, efficacy and safety results from the FD cohort are consistent with those from the MRD cohort,16,17 representing a robust dataset (n = 323) of patients treated with ibrutinib plus venetoclax in the CAPTIVATE study. FD combination treatment with ibrutinib plus venetoclax for 12 cycles provides clinically meaningful PFS and treatment-free remissions for patients with previously untreated CLL, including those with high-risk disease features; most patients remain progression-free 1 year after treatment.
Acknowledgments
The authors thank all the patients who participated in this study and their supportive families, as well as the investigators and clinical research staff from the study centers. The authors also thank Lisa J. Croner for her valuable contributions to bioinformatics analysis of next-generation sequencing data; Lisa J. Croner is an employee of AbbVie and owns AbbVie stock.
This study was supported by Pharmacyclics LLC, an AbbVie Company. Medical writing support was provided by Melanie Sweetlove and funded by Pharmacyclics LLC, an AbbVie Company.
Authorship
Contribution: C.S.T., J.N., J.P.D., C.Z., W.G.W., K.R., and P.G. designed the study; C.S.T., J.N.A., T.S., T.J.K., S.O., P.M.B., A.T., L.T., R.B., S.J., B.J.K., C.M., W.G.W., R.J., and P.G. enrolled patients and collected the data; C.Z. and E.S.-G. provided statistical data analysis; and all authors had access to the data and were involved in the interpretation of data, contributed to manuscript review and revisions, and approved the final version for submission.
Conflict-of-interest disclosure: C.S.T. reports honoraria from Janssen, AbbVie, BeiGene, Janssen, and Loxo Oncology; and research funding from AbbVie, BeiGene, and Janssen. J.N.A. reports honoraria from AbbVie, AstraZeneca, and Janssen; a consulting/advisory role for AbbVie, ADC Therapeutics, Ascentage, AstraZeneca, BeiGene, Epizyme, Genentech, TG Therapeutics, and Pharmacyclics LLC, an AbbVie Company; research funding from AstraZeneca, Celgene, Genentech, Janssen, and TG Therapeutics; and speakers bureau for AbbVie, AstraZeneca, BeiGene, Janssen, and Pharmacyclics LLC, an AbbVie Company. T.S. reports a consulting/advisory role for AstraZeneca, BeiGene, Bristol Myers Squibb, Celgene, Juno Therapeutics, and Kite Pharma; research funding from AstraZeneca, Ascentage, BeiGene, Celgene, Juno Therapeutics, Kite Pharma, Oncternal, TG Therapeutics, and Pharmacyclics LLC, an AbbVie Company; and speakers bureau for AstraZeneca, BeiGene, Janssen, and Pharmacyclics LLC, an AbbVie Company. T.J.K. reports a consulting/advisory role for AbbVie, Celgene, Genentech-Roche, Gilead Sciences, and Pharmacyclics LLC, an AbbVie Company; research funding from AbbVie, Genentech-Roche, Oncternal Therapeutics, and Pharmacyclics LLC, an AbbVie Company. R.J. reports a consulting/advisory role for AbbVie, AstraZeneca, Verastem, and Pharmacyclics LLC, an AbbVie Company; research funding from MEI Pharma, TeneoBio, TG Therapeutics, and Pharmacyclics LLC, an AbbVie Company; and speakers bureau for AbbVie, AstraZeneca, BeiGene, Janssen, and Pharmacyclics LLC, an AbbVie Company. S.O. reports honoraria from and a consulting/advisory role for AbbVie, Antengene, AstraZeneca, CSL, Gilead, Janssen, Merck, Roche, and Takeda; and research funding from AbbVie, Antengene, AstraZeneca, BeiGene, CSL, Gilead, Janssen, Roche, Merck, Takeda, and Pharmacyclics LLC, an AbbVie Company. P.M.B. reports a consulting/advisory role for AbbVie, AstraZeneca, Bristol Myers Squibb, Celgene, Genentech, Gilead, Janssen, Merck, MEI Pharma, MorphoSys, Seagen, TG Therapeutics, and Pharmacyclics LLC, an AbbVie Company; and research funding from AstraZeneca and TG Therapeutics. A.T. reports a consulting/advisory role and speakers bureau for AbbVie, AstraZeneca, BeiGene, and Janssen. L.T. reports honoraria from and a consulting/advisory role for AbbVie, Janssen, and Takeda; and research funding from and speakers bureau for Janssen. R.B. reports employment and stock or other ownership of an immediate family member with Sanofi-Pasteur; and research funding from AbbVie, Genentech-Roche, Merck, Regeneron, and Pharmacyclics LLC, an AbbVie Company. S.J. reports a consulting/advisory role and speakers bureau for AbbVie; research funding from AbbVie, GlaxoSmithKline, and Roche; and travel/accommodations/expenses from Roche. B.J.K. reports stock or other ownership with Commonwealth Serum Laboratories; honoraria from and a consulting/advisory role for AbbVie, Janssen, Kyowa Kirin, Merck, Mundipharma, Roche, and Takeda; speakers bureau for AbbVie, Janssen, and Roche; and expert testimony for AbbVie. C.M. reports a consulting/advisory role for AbbVie, AstraZeneca, BeiGene, and Janssen; and research funding from and speakers bureau for AbbVie and Janssen. E.S.-G., K.R., C.Z., and J.P.D. report employment with Pharmacyclics LLC, an AbbVie Company; and stock or other ownership with AbbVie. J.N. reports employment with Summit Therapeutics and previous employment with Pharmacyclics LLC, an AbbVie Company; and stock or other ownership with AbbVie. W.G.W. reports research funding from AbbVie, Acerta/AstraZeneca, Cyclacel, Genentech, Gilead Sciences, GlaxoSmithKline/Novartis, Janssen, Juno Therapeutics, Kite Pharma, Loxo Oncology, Miragen, Oncternal Therapeutics, Sunesis, Xencor, and Pharmacyclics LLC, an AbbVie Company. P.G. reports honoraria from and a consulting/advisory role for AbbVie, AstraZeneca, ArQule/MSD, Celgene/Juno/Bristol Myers Squibb, Janssen, Lilly/Loxo, MEI Pharma, and Roche; and research funding from AbbVie, AstraZeneca, Janssen, and Sunesis.
Correspondence: Constantine S. Tam, Peter MacCallum Cancer Center, 305 Grattan St, Melbourne, VIC 3000, Australia; e-mail: constantine.tam@petermac.org.
Requests for access to individual participant data from clinical studies conducted by Pharmacyclics LLC, an AbbVie Company, can be submitted through the Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal