

Key Points
Biallelic variants in HEATR3 are associated with DBA and other clinical features in humans.
HEATR3 variants destabilize the protein, resulting in a reduction of nuclear uL18 and impaired ribosome biogenesis.

Abstract
The congenital bone marrow failure syndrome Diamond-Blackfan anemia (DBA) is typically associated with variants in ribosomal protein (RP) genes impairing erythroid cell development. Here we report multiple individuals with biallelic HEATR3 variants exhibiting bone marrow failure, short stature, facial and acromelic dysmorphic features, and intellectual disability. These variants destabilize a protein whose yeast homolog is known to synchronize the nuclear import of RPs uL5 (RPL11) and uL18 (RPL5), which are both critical for producing ribosomal subunits and for stabilizing the p53 tumor suppressor when ribosome biogenesis is compromised. Expression of HEATR3 variants or repression of HEATR3 expression in primary cells, cell lines of various origins, and yeast models impairs growth, differentiation, pre–ribosomal RNA processing, and ribosomal subunit formation reminiscent of DBA models of large subunit RP gene variants. Consistent with a role of HEATR3 in RP import, HEATR3-depleted cells or patient-derived fibroblasts display reduced nuclear accumulation of uL18. Hematopoietic progenitor cells expressing HEATR3 variants or small-hairpin RNAs knocking down HEATR3 synthesis reveal abnormal acceleration of erythrocyte maturation coupled to severe proliferation defects that are independent of p53 activation. Our study uncovers a new pathophysiological mechanism leading to DBA driven by biallelic HEATR3 variants and the destabilization of a nuclear import protein important for ribosome biogenesis.
Introduction
Ribosomopathies are rare human disorders resulting from impaired production of ribosomes. Many variants in different ribosomal protein (RP) genes drive Diamond-Blackfan anemia (DBA). Rare cases have also been associated with the transcription factor GATA1 and the ribosomal assembly factor TSR2, whereas DBA-like syndromes were linked to variants of the genes encoding the adenosine deaminase ADA2 or the serum protein erythropoietin.1-4 DBA is characterized by a paucity of erythroid progenitor and precursor cells in the bone marrow and red cell aplasia that is frequently (∼50%) associated with physical malformations of the face, hands (abnormal thumbs), heart, and urogenital tract.5 Individuals with DBA also have an increased risk of developing cancer, including acute myeloid leukemia and osteosarcomas.6 Inherited RP gene mutations have been reported in individuals with asplenia, intellectual disability, autism, and/or dysmorphism but no hematologic phenotype.7-11 Suggested mechanisms underlying DBA include stabilization of the p53 tumor suppressor; the reduced translation of transcripts important for erythropoiesis such as GATA1, BAG1, and CSDE1; and/or the degradation of the GATA1 chaperone heat shock protein 70.1,12-17
RPL5 and RPL11 encode uL18 and uL5, respectively, which uniquely assemble in the nucleus with 5S ribosomal RNA (rRNA) to form the 5S ribonucleoprotein particle (RNP), a complex that incorporates with maturing large ribosomal subunits to form the central protuberance.18 When ribosome biogenesis is compromised and the 5S RNP is not incorporated, it accumulates in free form and associates with Hdm2. This interaction inhibits the Hdm2 ubiquitin ligase function that normally targets p53 for constitutive proteasomal degradation.19 This antitumor regulatory loop, known as nucleolar surveillance, regulates the p53 stabilization and apoptosis resulting from ribosome biogenesis inhibition.20 Importantly, all 3 components of the 5S RNP are dependent on each other to bind Hdm2, and, as such, reduced uL18 or uL5 in several models fails to induce p53 stabilization to the same extent as the loss of other RPs.21-24
Symportin 1 (Syo1, HEATR3 in human) is a yeast protein that coimports uL5 and uL18 into the nucleus.25,26 Syo1 associates cotranslationally with the N-terminus of uL18 and interacts with uL5, allowing for synchronized nuclear import of the newly synthesized RPs. Once in the nucleus, a pre–5S RNP is formed by the incorporation of 5S rRNA with Syo1-uL18-uL5, which is then recruited onto the pre-60S large ribosomal subunit.27 Although coimmunoprecipitation experiments in human cells have recently shown that HEATR3 associates with both uL5 and uL18,28 it remains unknown if the cotranslational capture is conserved.
The current study reports missense and splice site variants in HEATR3 (MIM 614951) in multiple individuals presenting with DBA and additional clinical features. Functional studies reveal defective ribosome biogenesis and reduced nuclear import of uL18/RPL5, and suggest that the impaired erythroid proliferation is independent of p53.
Methods
Affected individuals
The exome sequencing results from Family A were brought to the attention of the European Diamond-Blackfan anemia (EuroDBA) consortium. This led to EuroDBA members querying their respective registries and the identification of the other families (supplemental Materials and methods, available on the Blood Web site). Written informed consent was obtained from affected individuals and/or parents before inclusion in this study, which was performed in accordance with the ethical standards of the Declaration of Helsinki. All procedures in patient 3 (P3) were performed according to standards of institutional and national ethical boards (Hacettepe University: GO15/721, GO19/604). P4 and his family were registered in the French DBA registry (CNIL acceptance no. 911387, CCTIRS-no. 11.295, May 12, 2011).
Cell culture
Lymphoblastoid cell lines (LCLs) were derived from Epstein-Barr virus immortalization of peripheral mononuclear cells isolated from whole blood using Ficoll (GE Life Sciences) and grown in RPMI 1640 (Gibco) containing 10% or 15% fetal calf serum, 1% l-glutamine, and 1% penicillin/streptomycin. Fibroblasts were generated by using a skin biopsy from the hip while the individual was anesthetized for a bone marrow biopsy, and growth was maintained in Dulbecco’s modified Eagle medium (Gibco) containing 10% fetal calf serum. HeLa cells were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum (Gibco).
Erythroid cell culture assays
Erythroid cell culture assays were performed as previously described.16,17,29 Antibodies and stains for fluorescence-activated cell sorting analysis were PC7- or phycoerythrin (PE)-conjugated CD34 (Beckman Coulter), allophycocyanin-conjugated CD36 (BD Biosciences), allophycocyanin- or PE-conjugated Band 3 (kindly provided by Mohandas Narla, New York Blood Center or The International Blood Group Reference Laboratory), PE/Cy7–conjugated interleukin-3R (Miltenyi Biotech), PE/Cy7 (PE)-coupled GPA (Life Technologies), and 7AAD (Miltenyi Biotech). Fluorescence-activated cell sorting analysis was conducted on an Influx flow cytometer (BD Biosciences). Data were analyzed by using FCS 6 Express Flow cytometry, version 6.06.0040 (De Novo Software).
Results
Variants in HEATR3 drive hematologic phenotypes with additional clinical features
Two of three children from Family A, P1 and P2, presented with intellectual disability, bone marrow failure with marked erythroid lineage involvement, and short stature as well as some minor facial and acromelic findings such as synophrys, pes planus, hallux valgus, and brachydactyly (Table 1; Figure 1A-B). The elder sister, P1, died of osteosarcoma at age 20 years. Initial whole-exome sequencing (WES) of the parents and affected children pointed to 5 candidate genes with either homozygous or compound heterozygous variants (supplemental Table 1). HEATR3 (MIM 614951) was the most likely candidate due to its potential role in ribosome biogenesis.25HEATR3 (NM_182922) variant c.1751G>A; p.(Gly584Glu) was heterozygous in both parents and homozygous in P1 and P2. Subsequent Sanger sequencing DNA from whole blood of the unaffected child revealed a heterozygous HEATR3 variant. Based on the hematologic phenotype found in P1 and P2, all 5 candidate genes, including HEATR3, were reported to the EuroDBA.
The clinical features of the individuals in this study
| Feature | P1 | P2 | P3 | P4 | P5 | P6 |
|---|---|---|---|---|---|---|
| Clinical findings | ||||||
| Age at last visit | Died at age 20 y | 10 y | 17 y | 11 y | 9 y | 5 y |
| Sex | Female | Male | Male | Male | Female | Male |
| Ethnicity | Turkish | Turkish | Turkish | French | Dutch | Dutch |
| Country | The Netherlands | The Netherlands | Turkey | France | The Netherlands | The Netherlands |
| Family | A | A | B | C | D | D |
| Variant in HEATR3 (ENST00000299192) | c.1751G>A p.(Gly584Glu) homozygous | c.1751G>A p.(Gly584Glu) homozygous | c.1337G>A p.(Cys446Tyr) homozygous | c.399 + 1G>T p.? heterozygous and c.719C> T p.(Pro240Leu) heterozygous | c.400T>C p.(Cys134Arg) homozygous | c.400T>C p.(Cys134Arg) homozygous |
| Systemic findings | ||||||
| Hematologic findings at presentation | Anemia | Anemia | Anemia | Anemia | Anemia | Anemia and thrombocytopenia |
| Body mass index (normal 18.5-24.9 kg/m2) | 29 kg/m2 | 23.7 kg/m2 | 28.1 kg/m2 | 18.7 kg/m2 | 22.8 kg/m2 | 16.3 kg/m2 |
| Height | < –3 SD | < –1 SD | < –4 SD | < –1 SD (growth hormone therapy at 4 y, < –3 SD at 2 mo) | < –1 SD | 0 SD |
| Head circumference | NA | Normal (0 SD) | < –3 SD | NA | > +1 SD | >+1 SD |
| Skeletal findings | Brachydactyly of hands, distal tapering of fingers, pes planus, hallux valgus | Pes planus, hallux valgus of the left foot, bilateral hypoplasia of the fourth and fifth toes | Severe genu valgum (operated), pes planus, brachydactyly of hands, hypoplasia of toes, sandal gap, cubitis valgus | None | Congenital hip dysplasia, preaxial polydactyly of right hand (operated), clinodactyly of left thumb, brachydactyly of first metacarpals, distal tapering of fingers | Brachydactyly of hands |
| Intellectual disability/ developmental delay | Mild | Mild | Mild | None | Mild | None |
| Facial dysmorphism | Synophrys, hypertelorism, low-set ears | Coarse hair, straight eyebrows, synophrys, protruding ears, full cheeks, narrow mouth | Down-slanted palpebral fissures, mild ptosis of left eye, widow's peak, micrognathy, protruding ears | None | Straight eyebrows | Down-slanted palpebral fissures, low-set ears |
| Skin findings | None | Cutis marmorata | Multiple nevi, facial freckling | Multiple nevi | None | None |
| Echocardiogram | NA | NA | Aortic regurgitation, dysplastic aortic valves | Interatrial septal defect (ostium secundum) | Normal | NA |
| Gastrointestinal findings | None | None | None | Persistent diarrhea, colitis | None | None |
| Other findings | Obesity, PCOS, hypotonia | Truncal obesity, ENT problems | Truncal obesity | None | Obesity | None |
| History of RBC transfusion | Yes | No | Yes | Yes | Yes | Transient |
| Response to corticosteroid | No | NA | Yes | No | NA | NA |
| Current treatment | — | None | Corticosteroid | Transfusion and iron chelation | Transfusion and iron chelation | None |
| Clinical course | Died of osteosarcoma at age 20 y | Currently in good clinical condition | Currently in good clinical condition | Currently in good clinical condition | Currently in good clinical condition | Currently in good clinical condition |
| Family history | ||||||
| Consanguinity | First-degree cousins | First-degree cousins | First-degree cousins | No | Likely | Likely |
| Family history | Yes, sibling (P1) | Yes, sibling (P2) | Yes, sibling | None | Yes, sibling (P6) | Yes, sibling (P5) |
| Feature | P1 | P2 | P3 | P4 | P5 | P6 |
|---|---|---|---|---|---|---|
| Clinical findings | ||||||
| Age at last visit | Died at age 20 y | 10 y | 17 y | 11 y | 9 y | 5 y |
| Sex | Female | Male | Male | Male | Female | Male |
| Ethnicity | Turkish | Turkish | Turkish | French | Dutch | Dutch |
| Country | The Netherlands | The Netherlands | Turkey | France | The Netherlands | The Netherlands |
| Family | A | A | B | C | D | D |
| Variant in HEATR3 (ENST00000299192) | c.1751G>A p.(Gly584Glu) homozygous | c.1751G>A p.(Gly584Glu) homozygous | c.1337G>A p.(Cys446Tyr) homozygous | c.399 + 1G>T p.? heterozygous and c.719C> T p.(Pro240Leu) heterozygous | c.400T>C p.(Cys134Arg) homozygous | c.400T>C p.(Cys134Arg) homozygous |
| Systemic findings | ||||||
| Hematologic findings at presentation | Anemia | Anemia | Anemia | Anemia | Anemia | Anemia and thrombocytopenia |
| Body mass index (normal 18.5-24.9 kg/m2) | 29 kg/m2 | 23.7 kg/m2 | 28.1 kg/m2 | 18.7 kg/m2 | 22.8 kg/m2 | 16.3 kg/m2 |
| Height | < –3 SD | < –1 SD | < –4 SD | < –1 SD (growth hormone therapy at 4 y, < –3 SD at 2 mo) | < –1 SD | 0 SD |
| Head circumference | NA | Normal (0 SD) | < –3 SD | NA | > +1 SD | >+1 SD |
| Skeletal findings | Brachydactyly of hands, distal tapering of fingers, pes planus, hallux valgus | Pes planus, hallux valgus of the left foot, bilateral hypoplasia of the fourth and fifth toes | Severe genu valgum (operated), pes planus, brachydactyly of hands, hypoplasia of toes, sandal gap, cubitis valgus | None | Congenital hip dysplasia, preaxial polydactyly of right hand (operated), clinodactyly of left thumb, brachydactyly of first metacarpals, distal tapering of fingers | Brachydactyly of hands |
| Intellectual disability/ developmental delay | Mild | Mild | Mild | None | Mild | None |
| Facial dysmorphism | Synophrys, hypertelorism, low-set ears | Coarse hair, straight eyebrows, synophrys, protruding ears, full cheeks, narrow mouth | Down-slanted palpebral fissures, mild ptosis of left eye, widow's peak, micrognathy, protruding ears | None | Straight eyebrows | Down-slanted palpebral fissures, low-set ears |
| Skin findings | None | Cutis marmorata | Multiple nevi, facial freckling | Multiple nevi | None | None |
| Echocardiogram | NA | NA | Aortic regurgitation, dysplastic aortic valves | Interatrial septal defect (ostium secundum) | Normal | NA |
| Gastrointestinal findings | None | None | None | Persistent diarrhea, colitis | None | None |
| Other findings | Obesity, PCOS, hypotonia | Truncal obesity, ENT problems | Truncal obesity | None | Obesity | None |
| History of RBC transfusion | Yes | No | Yes | Yes | Yes | Transient |
| Response to corticosteroid | No | NA | Yes | No | NA | NA |
| Current treatment | — | None | Corticosteroid | Transfusion and iron chelation | Transfusion and iron chelation | None |
| Clinical course | Died of osteosarcoma at age 20 y | Currently in good clinical condition | Currently in good clinical condition | Currently in good clinical condition | Currently in good clinical condition | Currently in good clinical condition |
| Family history | ||||||
| Consanguinity | First-degree cousins | First-degree cousins | First-degree cousins | No | Likely | Likely |
| Family history | Yes, sibling (P1) | Yes, sibling (P2) | Yes, sibling | None | Yes, sibling (P6) | Yes, sibling (P5) |
ENT, ears, nose, throat; NA, not available; PCOS, polycystic ovarian syndrome; RBC, red blood cell; SD, standard deviation.
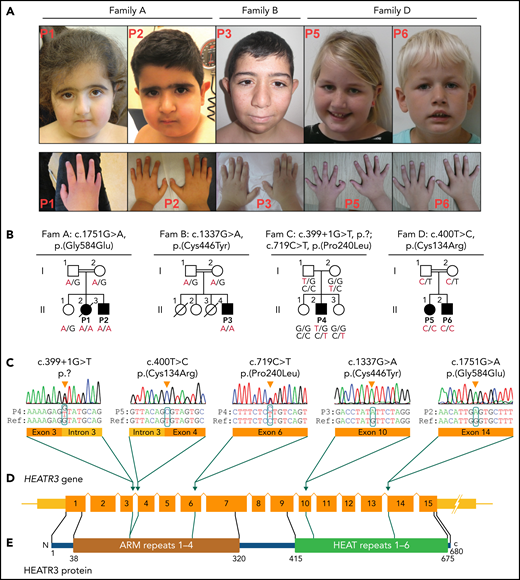
Clinical presentation and HEATR3 variants that drive DBA. (A) Photographs of the face and hands of the affected P1, P2, P3, P5, and P6 (photos from Family C not available). No common facial finding was present in affected individuals, but straight eyebrows, down-slanting palpebral fissures, and synophrys are apparent in some. Note that fingers in P1, P3, and P6 appear disproportionately short compared with the hand. Also note the presence of thumb anomaly in P5. (B) The pedigrees of Families A to D with genotypes for specified HEATR3 variants are indicated below each available individual. The pathogenic variants are indicated in red. (C) HEATR3 variants cosegregating with DBA in one affected individual from each family arranged according to position of coding sequence in ascending order. The missense variants c.400T>C (p.Cys134Arg) in P5, c.1337G>A (p.Cys446Tyr) in P3, and c.1751G>A (p.Gly584Glu) in P1 and P2 are homozygous; the splice donor site variant c.399 + 1G>T (p.?) and the missense variant c.719C>T (p.Pro240Leu) are compound heterozygous in P4. (D) Schematic representation of HEATR3 with 15 exons. (E) HEATR3 protein with four ARM (Armadillo) and six HEAT (Huntingtin, Elongation factor 3, protein phosphatase 2A, and Target of rapamycin 1) repeat domains, indicating the position of associated variants. The protein domains have been described previously.27,28
Clinical presentation and HEATR3 variants that drive DBA. (A) Photographs of the face and hands of the affected P1, P2, P3, P5, and P6 (photos from Family C not available). No common facial finding was present in affected individuals, but straight eyebrows, down-slanting palpebral fissures, and synophrys are apparent in some. Note that fingers in P1, P3, and P6 appear disproportionately short compared with the hand. Also note the presence of thumb anomaly in P5. (B) The pedigrees of Families A to D with genotypes for specified HEATR3 variants are indicated below each available individual. The pathogenic variants are indicated in red. (C) HEATR3 variants cosegregating with DBA in one affected individual from each family arranged according to position of coding sequence in ascending order. The missense variants c.400T>C (p.Cys134Arg) in P5, c.1337G>A (p.Cys446Tyr) in P3, and c.1751G>A (p.Gly584Glu) in P1 and P2 are homozygous; the splice donor site variant c.399 + 1G>T (p.?) and the missense variant c.719C>T (p.Pro240Leu) are compound heterozygous in P4. (D) Schematic representation of HEATR3 with 15 exons. (E) HEATR3 protein with four ARM (Armadillo) and six HEAT (Huntingtin, Elongation factor 3, protein phosphatase 2A, and Target of rapamycin 1) repeat domains, indicating the position of associated variants. The protein domains have been described previously.27,28
Queries into the Turkey EuroDBA registry revealed a second consanguineous family with an affected child and 3 siblings, 2 of whom died in early childhood (Family B). The affected child, P3, had intellectual disability, pure erythroid hypoplasia, short stature, and some minor dysmorphic findings overlapping with those of P1 and P2 (Table 1; Figure 1A-B). A focus on the 5 candidate genes in WES (supplemental Table 1) revealed a homozygous c.1337G>A; p.(Cys446Tyr) variant in HEATR3, supporting the findings in Family A. Similar screening of the French DBA registry identified a compound heterozygous variant in HEATR3 that coupled a canonical splice-site variant c.399 + 1G>T with a missense variant c.719C>T; p.(Pro240Leu), which segregate from either parent (Family C). In marked contrast to patients from Families A and B, the affected individual in Family C, P4, had no intellectual disability or dysmorphic findings but had short stature, pure erythroid hypoplasia, and interatrial septal defect (ostium secundum).
Finally, a fourth family of Dutch origin was identified with HEATR3 variants. The eldest sister (P5) presented with a severe anemia, intellectual disability, operated preaxial polydactyly in the right hand and clinodactyly of the left thumb, mild facial dysmorphic features, and congenital hip dysplasia (Table 1; Figure 1A-B). The brother (P6) had anemia and transient thrombocytopenia with mild dysmorphic facies and no intellectual disability. WES in P5 identified the homozygous c.400T>C; p.(Cys134Arg) variant in HEATR3. Sanger sequencing revealed this homozygous HEATR3 variant in P6, and a heterozygous variant was found in both parents. This variant is predicted to be “deleterious” according to SIFT and “disease-causing” by Mutation Taster but not predicted to affect splicing according to HSF, MaxEntScan, and NNSPLICE.30-34 None of these variants was found in the gnomAD, except for the c.400T>C; p.(Cys134Arg) variant in Family D, which is very rare with an allele frequency of 4.11 × 10−6 and not present in homozygosity.35
At the amino acid level, all these human variants occur at positions that are highly conserved evolutionarily (supplemental Figure 1). Sanger sequencing performed on DNA extracted from LCLs (in cases of P2 and P3), isolated lymphocytes (P4), and peripheral blood (P5 and P6) confirmed the expected HEATR3 variants (Figure 1C). These variants are found dispersed across HEATR3 with no apparent clustering except that they all affect residues in the ARM and HEAT repeat domains of the HEATR3 protein (Figure 1D-E). To exclude other biallelic genomic variants, common long contiguous stretches of homozygosity >5 Mbp were identified for individuals P1, P2, P3, and P5, which revealed a single common long contiguous stretch of homozygosity on chromosome 16 ([GRCh37] 25 263,278-53 191,470) where HEATR3 lies (supplemental Figure 2).
HEATR3 p.Gly584 (yeast p.Gly522) residue is important for protein function
In budding yeast, Syo1 is important for 60S ribosomal subunit assembly.25 Supplemental Figure 1 illustrates the amino acids affected by the 4 human variants, highlighting that only p.Gly584 has a homologous residue in yeast Syo1, p.Gly522. To test the effects of the p.Gly584 HEATR3 mutation, suitable yeast strains were engineered. Yeast cells with deletions in syo1 (syo1Δ) are viable but display a mild growth defect (apparent at 103× and higher dilutions in a plate assay) (Figure 2B) and a striking accumulation of 40S subunits (Figure 2A). The p.Gly584 HEATR3 residue was mutated in yeast Syo1 to yield p.(Gly522Ala) to probe function or p.(Gly522Glu) to mimic the variant in P1 and P2. Expression of the mutated constructs did not restore growth or the ribosomal subunit imbalance. By comparison, introduction of wild-type Syo1 complemented both growth and ribosomal subunit accumulation (Figure 2A-B). Western blot analysis revealed that neither variant had a noticeable impact on protein abundance (Figure 2C). Mapping the human p.Gly584 residue onto the Chaetomium thermophilum Syo1 three-dimensional structure, established in complex with uL18 by X-ray crystallography,25 revealed the presence of the equivalent p.Gly580 amino acid in an α-helix at a distance from the contacts with the RP (Figure 2D). This finding suggests that the pathogenicity of the HEATR3 p.(Gly584Glu) variant is linked to efficient 60S ribosomal subunit production.
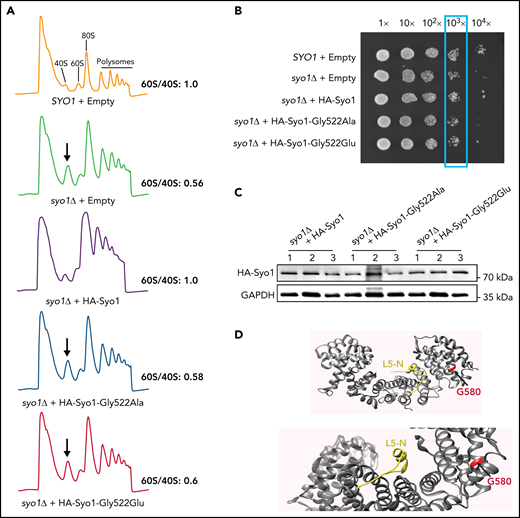
The HEATR3 Gly584 (yeast Gly522) residue is important for protein function. (A) Polysome profiles of wild-type and syo1Δ yeast cells expressing empty vector controls or HA-tagged Syo1-Gly522 mutations display an altered 60S/40S subunits ratio. Representative examples of a duplicate are shown. (B) Mutations at position Gly522 do not complement yeast growth. The mild growth impairment observed in syo1Δ cells is complemented by a wild-type construct (HA-Syo1) but not by constructs harboring a mutation at position Gly522. The indicated strains were grown at 16°C on synthetic medium lacking leucine for 6 days. (C) The Gly522 mutants are stably expressed in yeast. HA-Syo1 constructs were detected by western blotting with an anti-HA antibody. As loading control, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) probing was performed. (D) Position of yeast mutation Gly522 on the three-dimensional structure of C thermophilum Syo1 in complex with L5-N (based on PDB 4GMN25). Yeast Gly522 corresponds to Gly584 in the C-terminal (supplemental Figure 1). Two views are shown.
The HEATR3 Gly584 (yeast Gly522) residue is important for protein function. (A) Polysome profiles of wild-type and syo1Δ yeast cells expressing empty vector controls or HA-tagged Syo1-Gly522 mutations display an altered 60S/40S subunits ratio. Representative examples of a duplicate are shown. (B) Mutations at position Gly522 do not complement yeast growth. The mild growth impairment observed in syo1Δ cells is complemented by a wild-type construct (HA-Syo1) but not by constructs harboring a mutation at position Gly522. The indicated strains were grown at 16°C on synthetic medium lacking leucine for 6 days. (C) The Gly522 mutants are stably expressed in yeast. HA-Syo1 constructs were detected by western blotting with an anti-HA antibody. As loading control, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) probing was performed. (D) Position of yeast mutation Gly522 on the three-dimensional structure of C thermophilum Syo1 in complex with L5-N (based on PDB 4GMN25). Yeast Gly522 corresponds to Gly584 in the C-terminal (supplemental Figure 1). Two views are shown.
To establish if the cotranslational capture of uL18 by Syo125,26 is conserved in higher eukaryotes, we transiently expressed HA-tagged HEATR3 in HeLa cells, stalled ribosomes with cycloheximide, and performed an anti-HA affinity purification. The recovered material was tested for the presence of RPL5 messenger RNAs (mRNAs) (coding for uL18) by reverse transcription quantitative real-time polymerase chain reaction. Compared with an untagged HEATR3 control, RPL5 mRNA was found significantly enriched in HA-HEATR3 pellets (supplemental Figure 3). No cotranslational recruitment of RPL5 mRNA was observed when the HA-HEATR3 p.(Cys446Tyr) variant was expressed. We also tested for the presence of RPL10 mRNA coding for uL16, an RP that is not known to be chaperoned by Syo1.26 Consistently, we detected no signal in the HA-HEATR3 or in the untagged control pellets for RPL10. We concluded that the cotranslational capture mechanism used by yeast Syo1 to chaperone uL18 is conserved by HEATR3 in human cells and disrupted by the HEATR3 p.(Cys446Tyr) variant.
HEATR3 missense variants destabilize the HEATR3 protein and reduce nuclear uL18
To assess the effects of patient mutations on HEATR3 metabolic stability, protein levels were established by western blotting using lysates from LCLs derived from P2 and P3. The HEATR3 protein was barely detectable in either of the affected samples compared with the healthy controls (Figure 3A). To determine if this effect was due to the variants driving constitutive ubiquitin-mediated degradation of HEATR3 (similar to the fate of p53 in LCL basal conditions24), cells were exposed to the proteasome inhibitor MG-132 and blotted again for HEATR3. No restoration of HEATR3 levels was observed upon MG-132 treatment, in contrast to the clear and expected increase of p53 protein. This reduction of HEATR3 protein was not due to a loss of HEATR3 transcription, as real-time quantitative polymerase chain reaction analysis revealed equivalent levels of HEATR3 mRNA in cells from healthy and affected individuals (Figure 3B). These results suggest that the HEATR3 variants p.(Cys446Tyr) and p.(Gly584Glu) trigger the destabilization of the protein independently of the proteasome and without affecting HEATR3 mRNA levels.

HEATR3 variants affect protein levels and uL18 nuclear localization. (A) Representative western blot of lysates from LCLs derived from P2 and P3 that were either untreated or exposed to the proteasome inhibitor MG-132. Specific antibodies are used to detect the relative amounts of HEATR3. Protein p53 provides a positive control showing proteasome inhibition by MG-132. Actin detection was used as loading control. (B) Real-time polymerase chain reaction analysis of complementary DNAs generated from LCLs derived from a healthy control or from P2 and P3 using primers to measure HEATR3 mRNA levels. The mRNAs of genes 36B4, ACTB, and GAPDH were used as references. Data shown are the results of 6 biological replicate experiments. (C) Western blot of lysates from HeLa cells transfected with 2 different siRNAs targeting HEATR3 (#1 and #2) or a control scrambled siRNA (SCR) probed with antibodies against HEATR3. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) detection was used as loading control. (D) Wide-field fluorescence microscopy of HeLa cells transfected with SCR or HEATR3 siRNAs and stained with 4′,6-diamidino-2-phenylindole (DAPI) (to label the nucleus) and antibodies against uL18, shown at ×20 magnification. The insets show enlarged pictures of the areas framed by dotted lines. The arrowheads point to stained nucleoli, which are observed in control cells but not after HEATR3 knockdown. (E) Fibroblasts derived from a healthy control or P2, stained with DAPI and antibodies against uL18 or uL5. Scale bar, 10 μm. (F) Quantification of the number of fibroblasts revealing nuclear staining of uL18.
HEATR3 variants affect protein levels and uL18 nuclear localization. (A) Representative western blot of lysates from LCLs derived from P2 and P3 that were either untreated or exposed to the proteasome inhibitor MG-132. Specific antibodies are used to detect the relative amounts of HEATR3. Protein p53 provides a positive control showing proteasome inhibition by MG-132. Actin detection was used as loading control. (B) Real-time polymerase chain reaction analysis of complementary DNAs generated from LCLs derived from a healthy control or from P2 and P3 using primers to measure HEATR3 mRNA levels. The mRNAs of genes 36B4, ACTB, and GAPDH were used as references. Data shown are the results of 6 biological replicate experiments. (C) Western blot of lysates from HeLa cells transfected with 2 different siRNAs targeting HEATR3 (#1 and #2) or a control scrambled siRNA (SCR) probed with antibodies against HEATR3. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) detection was used as loading control. (D) Wide-field fluorescence microscopy of HeLa cells transfected with SCR or HEATR3 siRNAs and stained with 4′,6-diamidino-2-phenylindole (DAPI) (to label the nucleus) and antibodies against uL18, shown at ×20 magnification. The insets show enlarged pictures of the areas framed by dotted lines. The arrowheads point to stained nucleoli, which are observed in control cells but not after HEATR3 knockdown. (E) Fibroblasts derived from a healthy control or P2, stained with DAPI and antibodies against uL18 or uL5. Scale bar, 10 μm. (F) Quantification of the number of fibroblasts revealing nuclear staining of uL18.
To test if the uL18 nuclear import role of Syo125 is conserved in human cells, HEATR3 expression was knocked down with small interfering RNAs (siRNAs) in HeLa cells (Figure 3C) and stained with antibodies recognizing uL18. Figure 3D shows that in control cells, uL18 accumulated in the cytoplasm and the nucleus with a particularly strong enrichment in the nucleolus (where ribosome biogenesis initiates). Upon depletion of HEATR3, this enrichment of uL18 was lost in nearly all cells.
To confirm this finding in primary patient cells, dermal fibroblasts were obtained from P2. Figure 3E shows that nuclear staining of uL18 in fibroblasts expressing the HEATR3 variant p.(Gly584Glu) is reduced compared with control cells (Figure 3F). Staining the depleted HeLa cells (data not shown) or fibroblasts (Figure 3E-F) with antibodies recognizing uL5 (RPL11) did not reveal any appreciable differences. We thus concluded that HEATR3 is important for nuclear import of uL18 and that uL5 seems to be imported through additional mechanisms.
Cells carrying HEATR3 variants reveal strong pre-rRNA processing defects
Northern blot analysis of total RNA extracts compared pre-rRNA processing in LCLs generated from 3 affected individuals and 3 healthy control subjects. A striking common feature of the profiles of affected individuals was the accumulation of the 32S pre-rRNA, and to a lesser extent of the 12S pre-rRNA (internal transcribed spacer 2) (Figure 4A,C). Quantitative analyses of precursor-to-product ratios for the affected individuals relative to controls (Ratio Analysis of Multiple Precursors36) showed a strong increase of 32S/41S ratios and a decrease of 12S/32S ratios (Figure 4B). This suggests a delayed endonucleolytic cleavage at site 4 in the internal transcribed spacer 2, which separates the 5.8S and the 28S rRNAs on the primary transcript. These results are reminiscent of similar accumulations of 32S pre-rRNA in LCLs from RPL5+/mut and RPL11+/mut individuals presenting with DBA.37
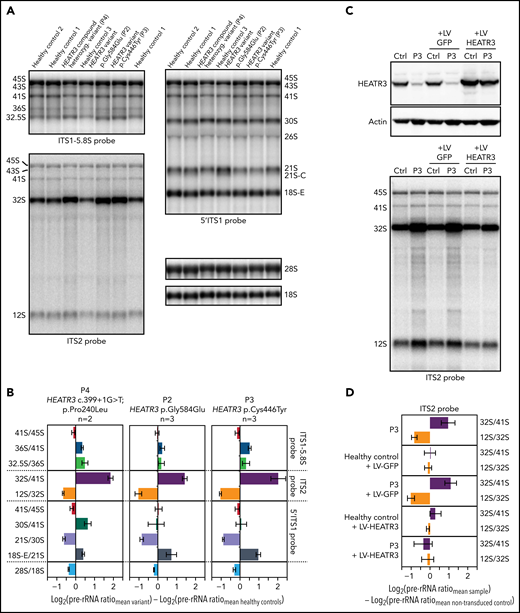
Variants in HEATR3 affect pre-rRNA processing. (A) Northern blot analysis of LCLs derived from control or affected individuals carrying HEATR3 variants. Radiolabeled probes targeting the 5′ extremity of internal transcribed spacer 1 (5′ITS1), the ITS1/5.8S junction (ITS1-5.8S), or internal transcribed spacer 2 (ITS2) were used to detect pre-rRNA precursors. Each lane was loaded with 3 µg total RNA. (B) Quantification of pre-rRNAs in LCLs derived from individuals carrying HEATR3 variants was performed by using Ratio Analysis of Multiple Precursors: product-to-precursor ratios at various processing steps are expressed as variations relative to LCLs from healthy controls. Mean values ± standard error of the mean from 3 independent experiments, aside from sample P4, for which n = 2. (C) Restoration of HEATR3 expression in LCLs from P3. LCLs from P3 and from a control individual were transduced with lentiviruses expressing either green fluorescent protein (LV-GFP) or HEATR3 (LV-HEATR3). Western blot analysis confirmed expression of HEATR3 in P3 LCLs transduced with LV-HEATR3 (upper panel). Northern blot analysis of total pre-rRNAs with probe ITS2 showed restoration of a normal pre-rRNA processing pattern. (D) Ratio Analysis of Multiple Precursors quantification of the northern blot shown in panel C. Product-to-precursor ratios in P3 LCLs at steps related to the processing of the large subunit rRNA precursors are expressed as variations relative to LCLs from the healthy control. Mean values ± standard error of the mean from 3 independent experiments.
Variants in HEATR3 affect pre-rRNA processing. (A) Northern blot analysis of LCLs derived from control or affected individuals carrying HEATR3 variants. Radiolabeled probes targeting the 5′ extremity of internal transcribed spacer 1 (5′ITS1), the ITS1/5.8S junction (ITS1-5.8S), or internal transcribed spacer 2 (ITS2) were used to detect pre-rRNA precursors. Each lane was loaded with 3 µg total RNA. (B) Quantification of pre-rRNAs in LCLs derived from individuals carrying HEATR3 variants was performed by using Ratio Analysis of Multiple Precursors: product-to-precursor ratios at various processing steps are expressed as variations relative to LCLs from healthy controls. Mean values ± standard error of the mean from 3 independent experiments, aside from sample P4, for which n = 2. (C) Restoration of HEATR3 expression in LCLs from P3. LCLs from P3 and from a control individual were transduced with lentiviruses expressing either green fluorescent protein (LV-GFP) or HEATR3 (LV-HEATR3). Western blot analysis confirmed expression of HEATR3 in P3 LCLs transduced with LV-HEATR3 (upper panel). Northern blot analysis of total pre-rRNAs with probe ITS2 showed restoration of a normal pre-rRNA processing pattern. (D) Ratio Analysis of Multiple Precursors quantification of the northern blot shown in panel C. Product-to-precursor ratios in P3 LCLs at steps related to the processing of the large subunit rRNA precursors are expressed as variations relative to LCLs from the healthy control. Mean values ± standard error of the mean from 3 independent experiments.
To prove that these processing defects are truly due to loss of HEATR3, we conducted complementation assays in LCLs. Expression of wild-type HEATR3 complementary DNA in patient LCLs (P3) by lentiviral infection was sufficient to restore normal pre-RNA processing (Figure 4C-D). In line with these results, acute depletion of HEATR3 in U2OS and HCT116 cells displayed similar changes in the pre-rRNA processing pattern (notably 32S accumulation) (supplemental Figure 4).
The levels of 60S ribosomal subunits are reduced in cells with HEATR3 variants
The impact of these pre-rRNA processing impairments on 60S ribosomal subunit formation was measured by subjecting the same LCLs to polysomal profiling. As in yeast, we found that LCLs carrying HEATR3 variants revealed a substantial loss of free 60S ribosomal subunits (Figure 5A-B). Also evident was a loss of 80S monosomes, which would be expected upon a 60S subunit shortage. These profiles are reminiscent of LCLs from individuals with DBA who carry variants in RPL genes.38-40 Expressing wild-type HEATR3 complementary DNA in patient LCLs (P3) with lentiviruses was sufficient to restore normal levels of 60S subunits (Figure 5B). Taken altogether, the results show that variants in HEATR3 have a major impact on the biogenesis of 60S ribosomal subunits.
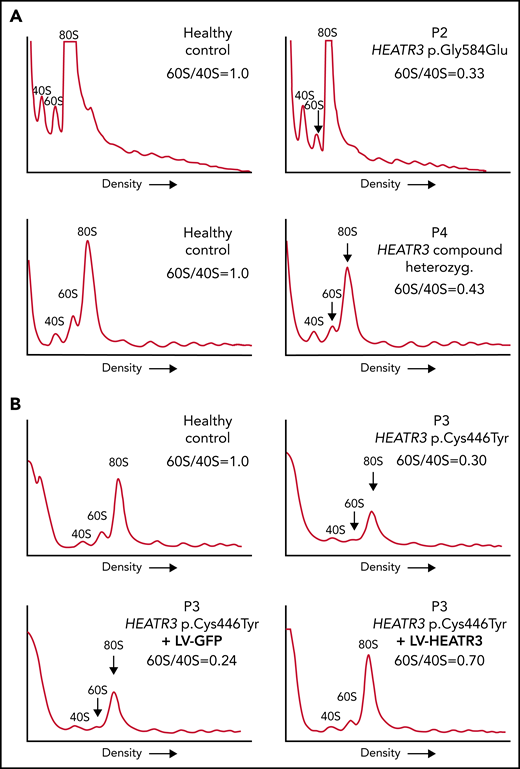
Variants in HEATR3 impair 60S ribosomal subunit accumulation. (A) Representative polysome profiles of lysates from LCLs derived from healthy individuals or patients (P2 and P4). The 40S subunit, 60S subunit, and 80S monosome are labeled. The vertical arrow highlights the reduction of 60S subunits in the samples from P2 and P4. (B) Representative polysome profiles of lysates from LCLs transduced with lentiviruses expressing HEATR3 or GFP complementary DNA. At least 3 independent experiments were performed with each cell line (panels A and B).
Variants in HEATR3 impair 60S ribosomal subunit accumulation. (A) Representative polysome profiles of lysates from LCLs derived from healthy individuals or patients (P2 and P4). The 40S subunit, 60S subunit, and 80S monosome are labeled. The vertical arrow highlights the reduction of 60S subunits in the samples from P2 and P4. (B) Representative polysome profiles of lysates from LCLs transduced with lentiviruses expressing HEATR3 or GFP complementary DNA. At least 3 independent experiments were performed with each cell line (panels A and B).
Variants or reductions in HEATR3 affect erythropoiesis
As part of a more complete hematologic evaluation, bone marrow smears of P2 and P5 were examined. The results revealed erythroblastopenia that was associated with hypolobulated megakaryocytes that retained a normal size, excluding dysmegakaryopoiesis (Figure 6A; Table 2). The erythroid cell cultures of CD34+ cells purified from bone marrow of P2 revealed severe defects in erythroid cell proliferation compared with a healthy control (Figure 6B). Comparing differentiation markers of P2 and healthy control cells in this assay by flow cytometry suggests that there were no detectable differences in erythroid precursor mortality (7AAD labeling) or in IL3Rneg, CD34neg/CD36+ expression levels at day 7 (supplemental Figure 5A). However, we observed a higher proportion of basophilic (both early and late) and orthophilic cells relative to proerythroblasts at days 10 and 12 in P2 cells, suggesting an acceleration of the terminal differentiation program (Figure 6C-D). This may be correlated to the increased Band 3 expression at day 10 in P2 compared with the healthy control (supplemental Figure 5B-C). A similar assay was performed with CD34+ cells purified from peripheral blood of P4, both parents of P4, and a healthy control subject. As with P2, a strong defect in erythroid cell proliferation was observed in the cells derived from P4 (supplemental Figure 5E).

Erythroid cell proliferation and differentiation are impaired in individuals with HEATR3 variants. (A) Bone marrow aspirates of P2 (left panels) and P5 (right panels) with May-Grünwald-Giemsa staining, demonstrating a paucity (arrows) or absence (P5) of erythroid precursors and hypolobulated megakaryocytes (lower panels). (B) Cell count of purified CD34+ from the peripheral blood of P2 (orange line) and a healthy control (blue line) subject to the erythroid culture assay was performed (n = 1 to minimize invasiveness). Shown are cell counts on days 5, 7, 10, and 12. (C) Flow cytometry analysis of differentiating erythroid cells from P2 and a healthy control at days 10 and 12. (D) Quantification of panel C. (E) Reverse transcription quantitative real-time polymerase chain reaction quantification of HEATR3 transcript levels in CD34+ cells infected with lentiviral constructs expressing shRNAs targeting HEATR3 or a scrambled control (SCR). (F) Cell numbers on days 5, 7, 10, and 12 of an erythroid culture assay after infection with the lentiviral vectors expressing HEATR3 shRNAs. (G) Western blot analysis of the cells on day 7. Blots were probed with antibodies against HEATR3, p21, eS19 (RPS19), heat shock protein 70 (HSP70), GATA1, and pro-caspase 3. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control. APC, allophycocyanin; EB, early basophilic erythroblasts; LB, late basophilic erythroblasts; ortho, acidophilic erythroblasts; poly, polychromatophilic erythroblasts; proE, proerythroblasts. **P < 0.002; ***P < 0.001; ****P < 0.0001.
Erythroid cell proliferation and differentiation are impaired in individuals with HEATR3 variants. (A) Bone marrow aspirates of P2 (left panels) and P5 (right panels) with May-Grünwald-Giemsa staining, demonstrating a paucity (arrows) or absence (P5) of erythroid precursors and hypolobulated megakaryocytes (lower panels). (B) Cell count of purified CD34+ from the peripheral blood of P2 (orange line) and a healthy control (blue line) subject to the erythroid culture assay was performed (n = 1 to minimize invasiveness). Shown are cell counts on days 5, 7, 10, and 12. (C) Flow cytometry analysis of differentiating erythroid cells from P2 and a healthy control at days 10 and 12. (D) Quantification of panel C. (E) Reverse transcription quantitative real-time polymerase chain reaction quantification of HEATR3 transcript levels in CD34+ cells infected with lentiviral constructs expressing shRNAs targeting HEATR3 or a scrambled control (SCR). (F) Cell numbers on days 5, 7, 10, and 12 of an erythroid culture assay after infection with the lentiviral vectors expressing HEATR3 shRNAs. (G) Western blot analysis of the cells on day 7. Blots were probed with antibodies against HEATR3, p21, eS19 (RPS19), heat shock protein 70 (HSP70), GATA1, and pro-caspase 3. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control. APC, allophycocyanin; EB, early basophilic erythroblasts; LB, late basophilic erythroblasts; ortho, acidophilic erythroblasts; poly, polychromatophilic erythroblasts; proE, proerythroblasts. **P < 0.002; ***P < 0.001; ****P < 0.0001.
Hematologic features of the individuals in this study
| P1 | P2 | P3 | P4 | P5 | P6 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hematologic findings | Reference values | First available | Most recent* | First available | Most recent | First available | Most recent | First available | Most recent | First available | Most recent | First available | Most recent |
| Blood parameters | |||||||||||||
| Hemoglobin | Age dependent (g/dL) | 6.8 | 7.2 | 10.5 | 10.3 | 7.4 | 10.1 | 3 | 8.1 | 4.5 | 7.1 | 5.6 | 9.4 |
| Reticulocytes | 25-120 (×109/L) | 108 | NA | 87.5 | 32.1 | NA | 96.9 | Decreased | 7.2 | NA | 14 | 127 | 38 |
| MCV | 80-97 fL | 84 | 86 | 81 | 78 | 88 | 94.8 | NA | NA | NA | 86.1 | NA | 80.4 |
| Leukocytes | 4.5-13 (×109/L) | 13.6 | 4.9 | 7.1 | 4.5 | 6.3 | 7.2 | Normal | 7.2 | 10.9 | 5.5 | 5.3 | 8.1 |
| Platelets | 150-450 (×109/L) | 373 | 76 | 239 | 213 | 235 | 248 | Normal | 220 | 269 | 288 | < 10 | 193 |
| HbF | <1% (after 1-2 y of age) | NA | NA | NA | 3.3 | NA | NA | NA | NA | NA | 1.0 | NA | 1.4 |
| eADA | <1.4 (U/g Hb) | NA | NA | 8.3 | NA | NA | 12.8 | NA | NA | NA | NA | NA | 4.02 |
| Bone marrow findings | |||||||||||||
| Erythroid lineage | Decreased | Decreased | Decreased | Decreased | Decreased | Decreased | |||||||
| Dyserythropoiesis | No | No | No | No | No | No | |||||||
| Megakaryocytic lineage | Normal | Normal | Normal | Normal | Normal | Normal | |||||||
| Remarks | Some monolobulated megakaryocytes, normal size | Some monolobulated megakaryocytes, normal size | |||||||||||
| P1 | P2 | P3 | P4 | P5 | P6 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hematologic findings | Reference values | First available | Most recent* | First available | Most recent | First available | Most recent | First available | Most recent | First available | Most recent | First available | Most recent |
| Blood parameters | |||||||||||||
| Hemoglobin | Age dependent (g/dL) | 6.8 | 7.2 | 10.5 | 10.3 | 7.4 | 10.1 | 3 | 8.1 | 4.5 | 7.1 | 5.6 | 9.4 |
| Reticulocytes | 25-120 (×109/L) | 108 | NA | 87.5 | 32.1 | NA | 96.9 | Decreased | 7.2 | NA | 14 | 127 | 38 |
| MCV | 80-97 fL | 84 | 86 | 81 | 78 | 88 | 94.8 | NA | NA | NA | 86.1 | NA | 80.4 |
| Leukocytes | 4.5-13 (×109/L) | 13.6 | 4.9 | 7.1 | 4.5 | 6.3 | 7.2 | Normal | 7.2 | 10.9 | 5.5 | 5.3 | 8.1 |
| Platelets | 150-450 (×109/L) | 373 | 76 | 239 | 213 | 235 | 248 | Normal | 220 | 269 | 288 | < 10 | 193 |
| HbF | <1% (after 1-2 y of age) | NA | NA | NA | 3.3 | NA | NA | NA | NA | NA | 1.0 | NA | 1.4 |
| eADA | <1.4 (U/g Hb) | NA | NA | 8.3 | NA | NA | 12.8 | NA | NA | NA | NA | NA | 4.02 |
| Bone marrow findings | |||||||||||||
| Erythroid lineage | Decreased | Decreased | Decreased | Decreased | Decreased | Decreased | |||||||
| Dyserythropoiesis | No | No | No | No | No | No | |||||||
| Megakaryocytic lineage | Normal | Normal | Normal | Normal | Normal | Normal | |||||||
| Remarks | Some monolobulated megakaryocytes, normal size | Some monolobulated megakaryocytes, normal size | |||||||||||
eADA, erythrocyte adenosine deaminase; Hb, hemoglobin; HbF, fetal hemoglobin; MCV, mean corpuscular volume; NA, not available.
During last phase of treatment for osteosarcoma.
To confirm the findings in Figure 6, erythroid cell culture assays were performed with CD34+ cells purified from cord blood and transduced with lentiviruses expressing small hairpin RNAs (shRNAs) targeting HEATR3 that reduced transcript levels (Figure 6E). CD34+ cells infected with these shRNAs displayed a dramatic decrease in erythroid proliferation compared with cells transduced with a scrambled control shRNA (Figure 6F). Although HEATR3 knockdown led to similar proportions of CD34+/CD36neg (erythroid burst-forming unit) cells, the increased proportion of CD34neg/CD36+ (erythroid colony-forming unit) cells at day 7 and day 10 compared with control (supplemental Figure 6) also suggests an acceleration of erythroid maturation. No apoptosis was observed; levels of the p53 target p21 and procaspase 3 proteins were unchanged (Figure 6G). No significant differences in expression of ILR3neg were detected (data not shown), and levels of eS19 (RPS19), heat shock protein 70, and GATA1 were similarly equivalent in control and HEATR3 shRNA-expressing cells. Some caution may be warranted in interpreting the western blot results given the different phases of maturation that the control vs HEATR3 knockdown cells reveal on the days collected for analysis.
HEATR3 variants do not alter levels of p53, uL5, or uL18 in LCLs
To confirm that p53 is not stabilized in cells with HEATR3 loss, western blot analysis was performed with lysates from patient LCLs. Blotting for p53 revealed that HEATR3 variants do not drive p53 stabilization (Figure 7A-B). Similarly, depletion of HEATR3 in U2OS or HCT116 cells also did not lead to p53 stabilization (supplemental Figure 4A). The p53 response to a DNA-damaging agent (camptothecin) or a proteasomal inhibitor (MG-132) was normal in patient LCLs. No difference was detected in the total levels of uL5 or uL18 in LCLs carrying HEATR3 variants (Figure 7C-D), suggesting that the nuclear reductions of uL18 observed in Figure 3 are not due to uL18 loss.
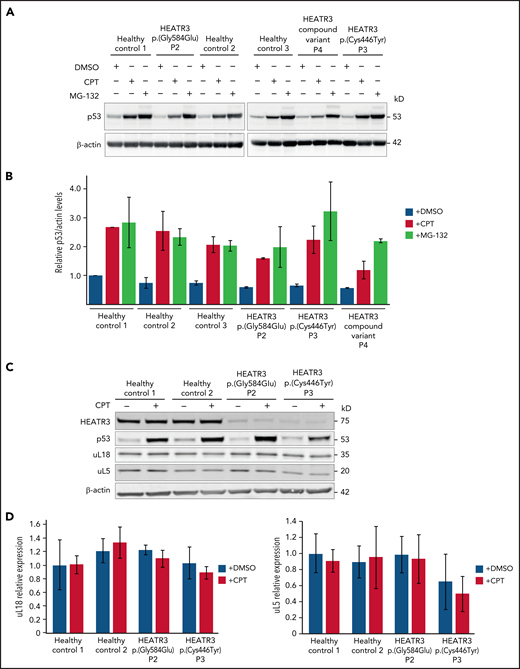
HEATR3 loss does not affect levels of p53, uL5, or uL18. (A) Western blots of LCLs exposed to either 100 nM camptothecin (CPT) for 4 hours, or 10 µM MG-132 for 6 hours, or the vehicle control, dimethyl sulfoxide (DMSO). Blots shown are probed with antibodies against p53. HEATR3 variants do not lead to p53 stabilization on their own but they do, as expected, in combination with CPT or MG-132 treatments. (B) Quantification of panel A from 3 independent experimental replicates. Results shown are the relative levels of p53/actin. (C) Western blots of LCLs exposed to 100 nM CPT for 4 hours, or the vehicle control, DMSO. Blots shown are probed with antibodies against HEATR3, p53, uL5, and uL18. (D) Quantification of panel C from 3 independent experimental replicates. Results shown are the relative levels of uL5/actin or uL18/actin.
HEATR3 loss does not affect levels of p53, uL5, or uL18. (A) Western blots of LCLs exposed to either 100 nM camptothecin (CPT) for 4 hours, or 10 µM MG-132 for 6 hours, or the vehicle control, dimethyl sulfoxide (DMSO). Blots shown are probed with antibodies against p53. HEATR3 variants do not lead to p53 stabilization on their own but they do, as expected, in combination with CPT or MG-132 treatments. (B) Quantification of panel A from 3 independent experimental replicates. Results shown are the relative levels of p53/actin. (C) Western blots of LCLs exposed to 100 nM CPT for 4 hours, or the vehicle control, DMSO. Blots shown are probed with antibodies against HEATR3, p53, uL5, and uL18. (D) Quantification of panel C from 3 independent experimental replicates. Results shown are the relative levels of uL5/actin or uL18/actin.
Discussion
We have identified HEATR3 as a novel causative gene of DBA in 4 distinct families with members carrying biallelic variants. Although DBA has been associated to date with at least 23 of the total 80 human RP genes,41 the number of non-RP genes driving this disorder remain far lower.1-4 We found that these HEATR3 variants drive DBA through a distinctive effect on ribosome biogenesis, whereupon reduced nuclear import of uL18 impairs production of 60S ribosomal subunits.
From a clinical perspective, the syndrome linked to HEATR3 mutations manifests as a variant form of DBA with additional features. Hematologically, the patients were all normocytic at diagnosis. Two patients were found to have thrombocytopenia at some stage, but it was transient in one patient (P6) and related to chemotherapy for osteosarcoma treatment in another (P1). Three of the 6 patients are transfusion dependent, 2 need no treatment at all, and 1 patient is treated with corticosteroids, suggesting that, in agreement with our general knowledge of DBA, the hematologic phenotype is heterogeneous, and patients may become treatment independent.42 Intellectual disability is rarely reported in classical DBA, and the few described cases are mostly linked to microdeletions that are considered to be driving a contiguous gene syndrome (unlike the individuals here).41 In contrast, other associated clinical features such as growth retardation (4 of 6 individuals) and acromelic dysmorphic features (5 of 6 individuals) are commonly reported in DBA.43 We observed a higher frequency of skeletal abnormalities (5 of 6 [83%]) and facial dysmorphism (5 of 6 [83%]) compared with congenital malformations in ∼50% of all patients with DBA. Interestingly, the abnormal thumb defect that is often reported in DBA patients with RPL5 variants37 is also observed in a majority of the patients with HEATR3 variants. In addition, 4 (60%) of 6 were diagnosed with (truncal) obesity, which was not treatment related in 3 patients. Of note, P4 reported unexplained severe diarrhea and colitis mimicking Crohn’s disease (Table 1). Interestingly, the HEATR3 allelic variation leading to the amino acid change p.(Arg642Ser) is reportedly associated with Crohn’s disease in the Ashkenazi Jewish population.44
All reported pathogenic ribosome-related gene variants in DBA are inherited dominantly.43,45 The only exception to this is the X-linked recessive hypomorphic variant in TSR2, a gene that codes for a pre-rRNA processing protein that contributes to ribosome biogenesis by promoting the assembly of RPS26/eS26.3HEATR3 is the second gene after TSR2 coding for a ribosome biogenesis factor linked to DBA, and, interestingly, the HEATR3 variants are also inherited in a recessive manner. Ribosome biogenesis factors are not integral components of the ribosomes; they interact transiently with maturing precursor subunits to promote numerous assembly reactions. Most of these trans-acting factors are essential, and some are thought to recycle upon function completion.46 Thus, ribosome biogenesis factors could be high-priority candidate genes for screening DBA families with recessive inheritance patterns.
The pathophysiology experiments reported here suggest that the variants p.(Cys446Tyr) and p.(Gly584Glu) impair the metabolic stability of the HEATR3 protein (Figure 3). This loss of protein very likely underlies the reduction of nuclear uL18 observed in cells with the p.(Gly584Glu) variant (Figure 3F) and in HeLa cells with knocked down HEATR3 (Figure 3C,E). This loss of uL18 accumulation in the nucleus is likely driving the impaired ribosome biogenesis observed in Figures 4 and 5. Despite this evident pre-rRNA processing defect, the ratio of mature 28S/18S rRNAs was only slightly affected in patient cells, in line with what was previously observed with DBA-linked RP gene variants.38,39 However, unlike knockdown of DBA-linked RPs, the mature rRNA ratio was also not severely affected upon severe depletion of HEATR3 in cell lines (supplemental Figure 4), indicating that HEATR3 is a facilitator of ribosome biogenesis but is not essential for the process. Similarly, Syo1 in yeast is dispensable for cell viability, and its deletion only marginally affects cell growth (Figure 2B). The perturbation resulting from HEATR3 loss in patient cells is nonetheless sufficient to cause a significant deficit of 60S subunits (Figure 5), which may alter the dynamics of translation and affect synthesis of proteins essential to erythroid differentiation.
Along the same line, an important finding of this work is that knocking down HEATR3 in CD34+ cells, or other cell lines, does not trigger p53 stabilization or upregulation of its target p21 (Figure 6G; supplemental Figure 4A). Accordingly, patient LCLs with HEATR3 variants showed no increase in basal p53 levels (Figure 7A-B), unlike what is observed in most LCLs derived from patients carrying RP gene variants.47 Indeed, nucleolar surveillance leading to p53 stabilization requires the nuclear accumulation of uL18,20 the precise step impaired by HEATR3 loss. Similar to our observations with HEATR3, RPL5 knockdown in cell lines does not stabilize p53, and LCLs from patients with DBA carrying RPL5 variants do not reveal increased levels of p53.20,47 This suggests a shared pathophysiological pathway between HEATR3 variants and one of the most commonly mutated RPL genes in DBA (RPL5). These in vitro data do not exclude a role for p53 in vivo, and future work is required for a full understanding of how HEATR3 deficiency leads to DBA.
One individual of this study (P1) died of osteosarcoma at age 20 years, suggesting that HEATR3 may be a tumor suppressor gene. This would agree with previous studies pointing to a tumor suppressor role for RPL5.48 Moreover, it has been reported that variants at the chromosomal locus 16q12.1 where HEATR3 resides have significant risk associations with several cancers, including glioblastoma (rs10852606, the risk allele “C” is associated with reduced HEATR3 expression), testicular (rs8046148 and rs10852606), and esophageal (rs4785204 and rs4785204) cancers.49-51 Further investigation is warranted, given the low number of individuals in this study.
We conclude that recessive loss-of-function variants in HEATR3 drive a pathogenic phenotype in humans that includes classical DBA with dysmorphic features and intellectual disability. This study shows that HEATR3 variants impair pre-rRNA processing and ribosome biogenesis by abrogating the cotranslational capture and nuclear import of uL18, an ancient and conserved function of the yeast ortholog Syo1. The hematologic defects linked to these variants seem to be caused by an abnormal acceleration of erythrocyte maturation coupled with a loss of proliferation capacity, and not by the stabilization of p53 or other known mechanisms driving DBA. This study adds a new disorder to the growing catalog of human ribosomopathies that drive blood disease.
Acknowledgments
The authors offer their most special thanks to the affected individuals, their families, and to the DBA patient associations. They thank all the people involved in the mutational screening analysis in France (Christine Bole and the Team from the Genomics Core Facility, Institut Imagine-Structure Fédérative de Recherche Necker, and technicians from the Hematology Laboratory in R. Debré Hospital, Paris), Turkey (Z. Ekim Taskiran for facilitating WES studies in Hacettepe University Exome Facility and Can Kosukcu for obtaining WES data), and to the registry data manager Isabelle Marie in France. They also thank Dirk Lebrecht and Marco Teller at the University Medical Center Freiburg for the generation of the LCLs and Patricia Veltman and Petra Mooyer at the Laboratory of Genetic Metabolic Disease at the Amsterdam University Medical Center for the generation of the fibroblasts. The authors are grateful for the expert assistance of Alexia Zakaroff and Elodie Riant (Genotoul-TRI, I2MC, Toulouse) to perform cell sorting and Frédéric Martins and Emeline Lhuillier (Genotoul-GET Santé, I2MC, Toulouse) for droplet digital polymerase chain reaction.
Specific E-Rare grants funding EuroDBA researchers are as follows: ZonMW #40-44000-98-1008 in the Netherlands (A.W.M.), TUBITAK 315S192 in Turkey (S.U.), and ANR-15-RAR3-0007-04 (P.-E.G., M.-F.O., and L.D.C.) in France. In addition, this project has received funding from the European Union’s Horizon 2020 research and innovation programme under the EJP RD COFUND-EJP No. 825575 to P.-E.G., L.D.C., D.L.J.L., R.H.H., and A.C. Specific grants under the frame of EJP RD programme JTC 2019 RiboEurope project are as follows: TUBITAK 319S062 in Turkey (A.C.), (EJP RD/JTC2019/PINT-MULTI) grant R.8015.19, and PDR grant T.0144.20 in Belgium (D.L.J.L.), as well as ANR-19-RAR4-0016 (P.-E.G.) and ANR-19-RAR4-0016-02 (L.D.C.) in France. P.-E.G., M.-F.O., and L.D.C. were also funded by ANR-2015-AAP générique-CE12 (DBA-Multigenes). L.D.C. is additionally supported by the Laboratory of Excellence for Red Cells [(LABEX GR-Ex)-ANR Avenir-11-LABX-0005-02], and The French National PHRC OFABD (DBA registry). D.L.J.L. is supported by the Belgian Fonds de la Recherche Scientifique (F.R.S./FNRS), the Université Libre de Bruxelles (ULB), the Région Wallonne (SPW EER) (“RIBOcancer” FSO grant 1810070, POC grant 1880014), the Fonds Jean Brachet, the Internationale Brachet Stiftung, and the Epitran COST action (CA16120). K.D.K. is supported by funding from the Foundation against Cancer (P2016-081 and 2016-112), and S.V. is a fellow at FWO (1S49817N).
Authorship
Contribution: S.U., P.O.S.-K., N.Y., A.C., N.A.A., L.D.C., M.B., E.B., K.v.G., M.S.-V., K.D.K., S.V., V.L., and T.L. were involved in the clinical care and molecular diagnosis of the individuals in the study; A.W.M., M.-F.O., L.D.C., M.B., P.-E.G., and D.L.J.L. designed the study; M.-F.O., M.L., C.J., M.S., S.D.L., L.W., N.L., E.L., N.M.-L., and J.P. designed and performed experiments; A.W.M., M.-F.O., L.D.C., M.L., C.J., R.H.H., P.-E.G., and D.L.J.L. analyzed and interpreted data; S.U., A.C., N.A.A., M.-F.O., L.D.C., M.B., K.D.K., V.L., P.-E.G., and D.L.J.L. contributed to the preparation and critical reviewing of manuscript; A.W.M. drafted the manuscript; and A.W.M., P.-E.G., and D.L.J.L. revised the manuscript. All authors approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Pierre-Emmanuel Gleizes, Centre de Biologie Intégrative, Université Paul Sabatier, 118 Route de Narbonne, 31062 Toulouse Cedex 9, France; e-mail: pierre-emmanuel.gleizes@univ-tlse3.fr; and Denis L. J. Lafontaine, Université libre de Bruxelles, Biopark Campus, Avenue des Professeurs Jeneer & Brachet 12, B-6041 Gosselies, Belgium; e-mail: denis.lafontaine@ulb.be.
Disease-causing HEATR3 variants have been submitted to ClinVar with the following accession numbers: SCV002003962, c.1751G>A p.(Gly584Glu), homozygous P1 and P2 ; SCV001983775, c.1337G>A p.(Cys446Tyr), homozygous P3 ; SCV002059206, c.399 + 1G>T p.? and SCV002059215, c.719C>T p.(Pro240Leu), compound heterozygous P4 ; SCV001976527, c.400T>C p.(Cys134Arg), homozygous P5 and P6. Data supporting the findings of this study are available upon request.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal