

In this issue of Blood, O’Donohue et al1 identify biallelic mutations in HEATR3as the underpinning cause of Diamond-Blackfan anemia (DBA) in 4 unrelated pedigrees. By using primary human cells, cell lines, and yeast models with HEATR3 deficiency, they delineate a mechanism by which reduced HEATR3 leads to erythroid failure. The mechanism includes impaired nuclear import of ribosomal protein uL18 (encoded by RPL5), defects in ribosomal RNA processing, and reduced production of the large (60S) ribosomal subunit, leading to p53-independent perturbation of erythroid development (see figure).
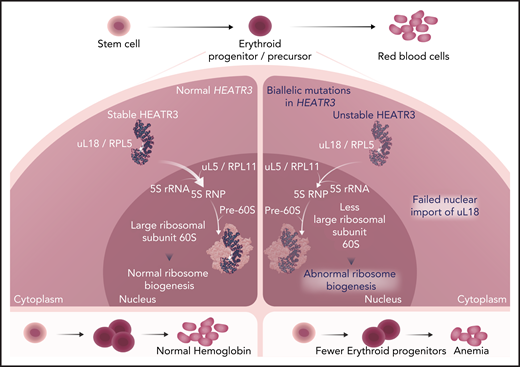
In normal bone marrow erythroid progenitors (left panel), HEATR3 acts as a shuttling factor that imports ribosomal protein uL18 from the cytoplasm to the nucleus. There, it associates with uL5 and the 5S ribosomal RNA (rRNA) to form the 5S ribonucleoprotein (RNP) complex, which is incorporated with maturing large ribosomal subunits to form the central protuberance. Intact ribosome biogenesis is a prerequisite for erythroid progenitor proliferation and differentiation to red blood cells. Biallelic HEATR3 variants destabilize the HEATR3 protein, reducing nuclear uL18 (right panel). HEATR3 variants cause pre-RNA processing defects, reduced 60S ribosomal subunits, and failure of erythropoiesis, presenting clinically as anemia. Professional illustration by Somersault18:24.
In normal bone marrow erythroid progenitors (left panel), HEATR3 acts as a shuttling factor that imports ribosomal protein uL18 from the cytoplasm to the nucleus. There, it associates with uL5 and the 5S ribosomal RNA (rRNA) to form the 5S ribonucleoprotein (RNP) complex, which is incorporated with maturing large ribosomal subunits to form the central protuberance. Intact ribosome biogenesis is a prerequisite for erythroid progenitor proliferation and differentiation to red blood cells. Biallelic HEATR3 variants destabilize the HEATR3 protein, reducing nuclear uL18 (right panel). HEATR3 variants cause pre-RNA processing defects, reduced 60S ribosomal subunits, and failure of erythropoiesis, presenting clinically as anemia. Professional illustration by Somersault18:24.
Genetic diagnosis of inherited bone marrow failure syndromes such as DBA is crucial for definitive diagnosis, screening of first-degree relatives and exclusion of asymptomatic carriers as bone marrow donors, and pre-implantation diagnosis. Approximately 75% of cases of DBA are caused by monoallelic loss-of function mutations in ribosomal protein (RP) genes.2 Fewer than 1% are associated with variants in the megakaryocyte-erythroid transcription factor GATA13 or the ribosome assembly factor TSR24; the mutations causing the remaining 20% to 25% of cases are unknown. In their work, O’Donohue et al elucidate the underlying pathogenetic mechanism in 6 individuals with DBA. Specifically, they used whole-exome sequencing and confirmatory Sanger sequencing to reveal homozygous or compound heterozygous variants in HEATR3, with distinct missense or splice site variants detected in each of the 4 families. The clinical phenotypes that are typical of DBA comprise selective erythroid hypoplasia (n = 5), anemia with transient but severe thrombocytopenia (n = 1), short stature (n = 5), facial dysmorphia and/or limb abnormalities (n = 5), congenital cardiac defects (n = 2), and intellectual disability (n = 4). One child died of osteosarcoma, one of the most common cancers arising in patients with DBA.5
As expected, missense variants depleted HEATR3 protein but not messenger RNA. To understand the mechanisms by which mutations in HEATR3 lead to a ribosomopathy, the authors exploited the homology between 1 of the 4 identified variants affecting amino acid p.Gly584 and residue p.Gly522 of yeast Syo1, important for 60S ribosomal subunit assembly. Complementation of Syo1-null yeast cells with a DBA-related variant failed to restore growth or ribosomal subunit imbalance. By contrast, wild-type Syo1 rescued both these defects. Similarly, in lymphoblastoid cell lines (LCLs) derived from 2 of the DBA patients, the 60S:40S imbalance and pre-RNA processing defects were corrected by lentiviral transduction of wild-type HEATR3 but not by green fluorescent protein control. These data suggest that the pathogenicity of the HEATR3 variants is related to efficient 60S subunit production. Syo1 facilitates nuclear translocation of a uL18-uL5 complex (see figure) through co-translational capture of uL18, and the authors show conservation of this mechanism in human cells. Furthermore, by using fluorescence microscopy of HEATR3-depleted cells (RNA interference–treated HeLa cells or primary patient skin fibroblasts) compared with controls, the authors concluded that HEATR3 is important for nuclear import of uL18 but not uL5. The reasons for this discrepancy are not established, but uL5 may be recruited to the HEATR3-uL18 complex in the nucleus or through a HEATR3-independent mechanism.
Consistent with the anemia affecting these patients, O’Donohue et al show that CD34+ cells derived from patients’ peripheral blood or normal CD34+ cells subjected to small hairpin RNA–mediated HEATR3 knockdown fail to expand in an erythroid culture assay. In addition, they exhibit accelerated erythroid maturation and normal GATA1 expression, concordant with the recent observation of earlier acquisition of erythroid differentiation markers and preserved GATA1 expression in bone marrow erythroid progenitors harvested from patients with DBA caused by mutations in RPL5 and RPL11.6 Finally, the article by O’Donohue et al demonstrates that HEATR3-depleted patient LCLs have comparable total cellular levels of uL18, consistent with the defect in uL18 subcellular localization, and they express similar levels of p53 compared with control LCLs. A link between haploinsufficiency of small ribosomal subunit proteins and p53 stabilization is well established because of the excess large subunit proteins which sequester HDM2 that can no longer bind to and promote proteasomal-mediated degradation of p53.7 The role of p53 in DBA caused by large subunit mutations is more contentious. Although RPL5-deficient embryonic stem cells demonstrate p53-independent growth defects,8 in line with the findings presented by O’Donohue, other data show enrichment of p53 and its molecular targets in RPL5- or RPL11-mutant DBA patient cells.6 This dichotomy implies a putative role for the bone marrow microenvironment and non-erythroid cells in modulating p53 expression. Further work is needed to discern how HEATR3 deficiency leads to erythroid failure in vivo.
So, what are the clinical implications of the findings of O’Donohue et al? HEATR3 should be included in multigene panels used for diagnosing DBA, particularly in the context of consanguinity (present in 3 of the 4 families studied). More broadly, this work demonstrates the value of international collaboration in a rare disease; after elucidation of HEATR3 variants in 1 family, the EuroDBA consortium helped identify 3 additional families with the same genetic basis.
To date, variants in only 1 other gene (TSR2) have demonstrated autosomal recessive inheritance in DBA. TSR2 acts as a nuclear unloading chaperone for RPS26 to ensure its safe transfer to maturing pre-ribosomes4, thus mediating a critical role in ribosome biogenesis, like HEATR3. Taken together, these data suggest that genetically unexplained DBA cases are likely attributable to defects in ribosome structure, assembly, or function.
One caveat is that although HEATR3 is depleted in all 6 patients, their clinical phenotype is heterogeneous and encompasses transfusion dependence, steroid responsiveness, and treatment independence. Although previous studies have elucidated some genotype-phenotype associations in DBA,6,9 more work is required to reveal the genetic, epigenetic, and environmental factors underpinning variable expressivity, in the hope that these could be harnessed for new therapies.
Although there was no evidence for p53 activation downstream of HEATR3 loss, notably the craniofacial defects arising in Treacher-Collins syndrome, which resemble those observed in patients with RPL5 or RP11 DBA9 as well as the patients in the study by O’Donohue et al, are caused by sensitization of neural crest cells to p53-mediated apoptosis.10 Most DBA research has focused on elucidating the mechanisms of anemia. However, delineating the relationship between ribosome defects and nonhematologic manifestations, such as the congenital anomalies, intellectual disability, and cancer reported in the study by O’Donohue et al, is essential for the holistic management of this complex disease and improving the quality of life of affected individuals.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal