

In this issue of Blood, Dinauer et al used NADPH oxidase 2 (NOX2)-deficient mice to study the underlying mechanisms of the noninfectious inflammatory conditions that arise in patients with chronic granulomatous disease (CGD).1 They identified the development of a new population of alveolar macrophages (AMs) with significant inflammatory properties.
After fetal liver monocytes populate each of the organs during development, they receive important instructions from the local environment to develop into noninflammatory, tolerant macrophages that regulate homeostasis. For example, in the liver, the monocyte reaches out and touches hepatocytes, stellate cells, and sinusoidal endothelium and, through key molecules that activate specific transcription factors, acquires its Kupffer cell identity.2 In the liver, these Kupffer cells sustain their number through replication. In other organs, such as the lung, the AMs are slowly replaced by monocyte-derived macrophages that are rapidly trained to take on the identity of original tissue-resident macrophages. Because only 30% to 40% of alveoli contain AMs at any one time, the cells migrate from alveolus to alveolus, ensuring that there is full coverage of all alveoli within the lung so that debris and any inhaled pathogens are removed.3 Despite the fact that we breath in 10 000 L of nonsterilized air each day, AMs are so efficient, that neutrophils are generally not needed. The small increase in bacterial burden is handled by the AMs through protective mechanisms, such as phagocytosis and the release of reactive oxygen species. Patients with CGD lack NOX. They develop infections and have increased inflammation, especially in the lungs. Clearly, an efficient offense is the best defense against inhaled foreign particles, and a perturbation in this well-oiled immune machine, such as loss of the oxidant-generating enzyme NOX2, causes a shift away from homeostasis.
Dinauer and colleagues unveil what appears to be an anti-inflammatory role for the bacteria-killing enzyme NOX2 in regulating sentinel AMs to ensure measured responses to disturbances, thereby maintaining pulmonary homeostasis. In NOX2 deficiency, neutrophil recruitment was noted in the absence of infection (see figure). Neutrophil recruitment into lungs is almost always associated with excessive inflammation that can damage bystander tissues and cells. However, the healthy AM is trained to remove even 1 million bacteria instilled directly into mouse lungs without a need for neutrophils. It is only when an overwhelming infection occurs that blood-borne immune cells are recruited.4 Because NOX2-deficient mice have lost their capacity to maintain homeostasis, neutrophils are inadvertently recruited, even at steady state. NOX2-deficient neutrophils may also contribute to the increased inflammation. Indeed, the global NOX2-knockout mice have greater inflammation than the macrophage-selective NOX2-knockout mice, suggesting that other cells, such as the neutrophils, also contribute to the inflammation and perhaps are even more inflammatory.
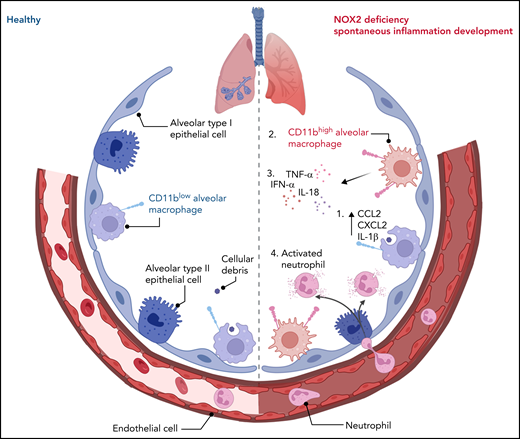
A healthy and NOX2-deficient alveolus. A healthy alveolus populated with sentinel resident CD11blow AMs that can capture cellular debris to maintain homeostasis (left). NOX2 deficiency results in (1) phagocytic CD11blow AMs that can capture cellular debris but also release production of proinflammatory cytokines and chemokines, followed by the emergence of (2) a CD11bhigh AM population, derived from either resident AMs or recruited monocytes, and by overall lung inflammation in mice and humans, characterized by (3) an increase in proinflammatory cytokines and (4) recruitment and activation of neutrophils (right). IFN-α, interferon-α; IL-18, interleukin-18; TNF-α, tumor necrosis factor-α.
A healthy and NOX2-deficient alveolus. A healthy alveolus populated with sentinel resident CD11blow AMs that can capture cellular debris to maintain homeostasis (left). NOX2 deficiency results in (1) phagocytic CD11blow AMs that can capture cellular debris but also release production of proinflammatory cytokines and chemokines, followed by the emergence of (2) a CD11bhigh AM population, derived from either resident AMs or recruited monocytes, and by overall lung inflammation in mice and humans, characterized by (3) an increase in proinflammatory cytokines and (4) recruitment and activation of neutrophils (right). IFN-α, interferon-α; IL-18, interleukin-18; TNF-α, tumor necrosis factor-α.
Dinauer et al have also uncovered a new population of CD11bhigh AMs within the CGD-affected mouse lung. Whether it is the loss of NADPH oxidase per se or the increased inflammation that derives this population is unclear. It has been reported that inflammation in the lung, triggered by external infectious pathogens also drives an increase in CD11bhigh AMs that is transcriptionally and epigenetically distinct from the resident CD11blow AMs.5 Comparably, using RNA sequencing and ATAC sequencing, Dinauer et al observed the greatest expression of proinflammatory genes in CD11bhigh AMs compared with wild-type AMs, suggesting a contribution of CD11bhigh AMs to the lung proinflammatory landscape. They did not actively examine the source of these cells; both monocyte-derived and tissue-resident macrophages could be contributors to this population. It is nevertheless intriguing that NOX2-deficient macrophages increased CCL2 production 250-fold in bronchoalveolar lavage fluid in response to lipopolysaccharide, a chemokine absolutely critical for monocyte mobilization from the bone marrow and recruitment into tissues.6,7 A monocyte-derived, cell-specific effect has also been shown in the peritoneum, where NOX2 absence delayed the maturation of monocyte-derived macrophages after zymosan instillation, resulting in a persistent inflammatory phenotype.8 Although Dinauer et al noted no increased apoptosis or decreased replication in the resident AMs, monocyte-derived macrophages could simply be added to the AM pool. Although they have been careful not to overinterpret the origin of these CD11bhigh AMs, a future monocyte lineage-tracing experiment using Ms4a3-RFP mice would clarify the extent of monocyte-derived macrophages on a NOX background.9 It would unveil whether the resident macrophages or monocyte-derived macrophages are being maladaptively trained in the absence of NOX2.
Although there is always criticism levied against inferring human phenotypes from mouse experiments, in this case the mouse largely corresponds to the human clinical condition. There are some notable differences; for example, whereas humans with CGD develop opportunistic infections such as Aspergillus fumigatus,10 mice remain reasonably devoid of such infections. However, this is almost certainly not a mouse model problem but an environmental problem. Mice kept in pathogen-free conditions are often devoid of many phenotypes found in humans living in the outside pathogen-laden world. Indeed, it would be fascinating to examine the CGD mouse in the wild to determine the full extent of the inflammatory phenotype in this deficiency. Although the germ-free environment did not unveil any difference in specific-pathogen–free mice, we predict that placing the mice in their natural environment and exposing them to mouse fungal, bacterial, and viral pathogens would further exacerbate the phenotype seen in this study. Indeed, exposing the NOX2-deficient mice to zymosan and Toll-like receptor ligands elicited more potent inflammatory responses than in wild-type mice. So, an ongoing challenge with pathogens and microbial products may further replicate the human condition.
The vulnerability to opportunistic infections and an increased inclination toward a proinflammatory landscape illustrates an extremely dangerous lung profile in patients with CGD. Dinauer et al inform us of the NOX2 absence–induced, altered-AM phenotype and its association with the spontaneous development of noninfectious inflammation in the lung. This study is a notable contribution to our understanding of the CGD lung and reminds us that the best immune defense is a very effective offense.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal