

In this issue of Blood, Farley et al1 use a mouse model of fetal and neonatal alloimmune thrombocytopenia (FNAIT) to show how risk of intracranial hemorrhage (ICH) varies with platelet count and developmental age. In addition to delineating critical platelet thresholds, they demonstrate that mice develop resilience to thrombocytopenia-associated ICH shortly after birth.
FNAIT results from placental transfer of maternal alloantibodies (IgGs) directed against paternally inherited antigens present on fetal platelets but absent from maternal platelets.2,3 These alloantibodies trigger accelerated clearance of platelets from the fetal and neonatal circulation. Hence, this condition is the platelet counterpart of hemolytic disease of the newborn. FNAIT occurs in ≈1 of every 1000 live births and is 1 of the principal causes of severe thrombocytopenia (platelet count <25 × 109/L) in fetuses and term newborns.2-4 Notably, approximately 15% of affected neonates have ICH (see figure panel A), and 50% of these cases occur antenatally. Among the long-term consequences of ICH in the ante- or neonatal period are hydrocephalus, porencephaly, seizures, and fetal demise. Treatment of FNAIT includes antenatal IV IgG to suppress platelet destruction and postnatal platelet transfusion.2,3
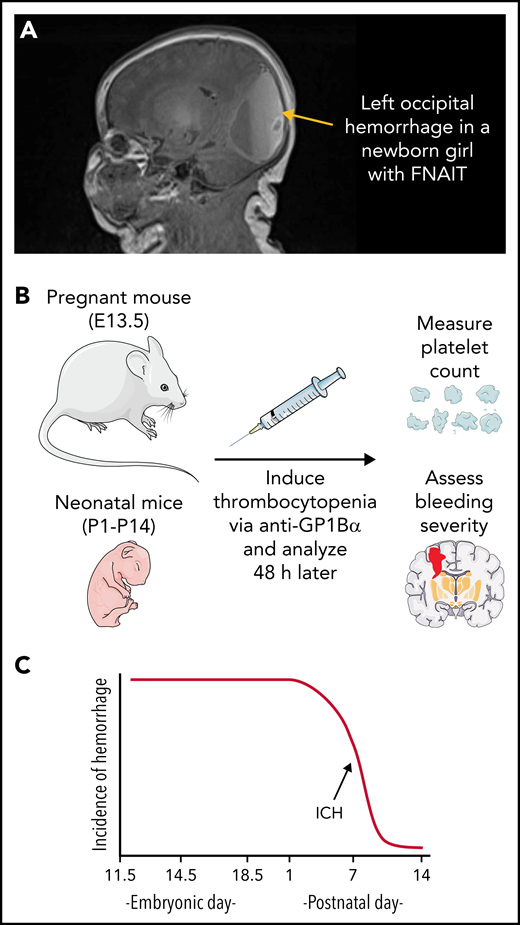
Fetal and neonatal alloimmune thrombocytopenia (FNAIT). (A) Magnetic resonance image (MRI) of a newborn girl with FNAIT caused by anti–HPA-3a. Note the large left occipital hemorrhage containing layering blood of different ages. Hemorrhage was identified on day of birth via ultrasound and confirmed on day of life 1 with MRI showing hemorrhage in the bilateral occipital lobes, left parietal lobe, and right temporal lobe. (B) Overview of the mouse model of FNAIT used by Farley et al. (C) Development of resilience to thrombocytopenia-induced ICH. Cartoons were prepared using adaptations of image vectors from Servier Medical Art (www.servier.com), licensed under the Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0).
Fetal and neonatal alloimmune thrombocytopenia (FNAIT). (A) Magnetic resonance image (MRI) of a newborn girl with FNAIT caused by anti–HPA-3a. Note the large left occipital hemorrhage containing layering blood of different ages. Hemorrhage was identified on day of birth via ultrasound and confirmed on day of life 1 with MRI showing hemorrhage in the bilateral occipital lobes, left parietal lobe, and right temporal lobe. (B) Overview of the mouse model of FNAIT used by Farley et al. (C) Development of resilience to thrombocytopenia-induced ICH. Cartoons were prepared using adaptations of image vectors from Servier Medical Art (www.servier.com), licensed under the Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0).
Currently, there are more than 30 known human platelet alloantigens (HPAs) expressed on surface glycoprotein (GP) complexes including GPIIb/IIIa (fibrinogen receptor), GP1b-V-IX (von Willebrand factor receptor), and GP1a/IIa (collagen receptor).2,3 In populations of European ancestry, the alloantigen most frequently implicated in FNAIT is HPA-1a, an epitope on GPIIIa, accounting for about 80% of cases. GPIIIa is expressed not only on platelets but also placental syncytiotrophoblasts exposed to the maternal circulation; therefore, HPA-1a alloimmunization may occur during a first pregnancy. Incompatibility for HPA-5b, a GPIa epitope common in African populations, is the next most frequent cause of FNAIT.
The overall incidence of FNAIT is much lower than predicted based on the distribution of alleles in the population; only 10% of HPA-1a–incompatible pregnancies result in maternal HPA-1a sensitization.4,5 Other factors, including immune response genes, impact alloantibody formation. For example, there is a strong association between HPA-1a alloantibody formation and HLA class II DRB3*01:01, which facilitates optimal antigen presentation to T cells.2 Bacterial or viral infections have been shown to augment the maternal alloimmune response to platelet antigens, increasing the severity of FNAIT in fetal mice.2,3 Although maternal risk factors are known, prenatal FNAIT risk assessment is not performed routinely, and most cases are diagnosed only after delivery.
In an earlier report, Farley et al5 induced thrombocytopenia in fetal or neonatal mice via administration of anti-GP1Bα and concluded that severe thrombocytopenia in utero or in the neonate was sufficient to cause ICH. Additionally, they observed a relationship between developmental stage and anatomic location of hemorrhage, with ICH most frequently occurring in the ganglionic eminence when thrombocytopenia was induced between embryonic day (E)10.5 and E12.5, and the location of bleeding shifting to the cortex and later the cerebellum when thrombocytopenia was induced between E14.5 and postnatal day (P)1. Now they have fine-tuned their model to address 2 key questions in the field of FNAIT research. (1) What platelet count threshold confers ICH risk? (2) What is the duration of neonatal susceptibility to thrombocytopenia-induced ICH? They injected varying amounts of anti-GP1Bα into pregnant mice at E13.5 (equivalent to 6-20 weeks of human development) and then analyzed the fetuses 48 hours later, measuring platelet counts and scoring hemorrhage severity (see figure panel B). A platelet count of ≥60% of normal prevented hemorrhage. At a platelet count of 30% of normal, 60% of the fetuses developed ICH. Below a platelet count of 10% of normal, all the mice developed ICH. To define the developmental risk window, severe thrombocytopenia (platelet count <5% of normal) was induced at P1 (equivalent to 23-32 weeks of human development), P7, or P14 (equivalent to 36-40 weeks of human development). All the mice injected at P1 developed ICH, whereas none of the mice injected at P14 did, implying that resilience to thrombocytopenia-induced ICH develops within the first 2 weeks of mouse life (roughly the equivalent of full term in humans; see figure panel C). These results, coupled with earlier studies,5 establish the following: (1) platelets are crucial for maintaining cerebrovascular integrity during prenatal and early neonatal development, and (2) the developmental stage of a thrombocytopenic insult is relevant to both the anatomic location and severity of injury.
The current work of Farley et al suggests a diminished risk for ICH in the setting of thrombocytopenia shortly after birth, but questions remain. Prior studies have shown an interaction between ongoing inflammation, thrombocytopenia, and bleeding risk in the adult mouse,6 leading one to wonder how inflammation may impact the platelet thresholds and developmental risk window defined in this work. What underlies the acquisition of resilience in this model? The authors speculate that platelets could limit bleeding associated with developmental vascular remodeling, as occurs during formation of mouse mesentery.7 In that system, activated platelets maintain vascular integrity by extending filopodia at sites of gaps between endothelial cells. Do cell intrinsic changes in thrombopoiesis contribute to the development of resilience? This is the identical timeline for when murine hematopoietic stem cells undergo transcriptional reprogramming from fetal-like to adult-like.8 How relevant are findings in this mouse model to FNAIT in humans? Although rodents are widely used as experimental models of neonatal brain injury, species differences in the timing of brain maturation can make comparisons of injury susceptibility difficult to interpret.9 The experimental system of Farley et al is based on anti-GP1Bα, yet most cases of human FNAIT are caused by antibodies directed against GPIIb/IIIa.2,3 Moreover, anti-GP1Bα causes both platelet clearance and perturbed GP1Bα signaling, which may enhance the severity of ICH in this model.5 These caveats notwithstanding, this mouse model may prompt clinicians to thoughtfully consider transfusion practices in neonates with FNAIT given the potential adverse clinical outcomes associated with unneeded platelet transfusions.10
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal