

Key Points
CRLF3 deficiency causes an isolated and sustained reduction in platelet count in mice.
CRLF3 is a potential therapeutic target for thrombocythemia.

Abstract
The process of platelet production has so far been understood to be a 2-stage process: megakaryocyte maturation from hematopoietic stem cells followed by proplatelet formation, with each phase regulating the peripheral blood platelet count. Proplatelet formation releases into the bloodstream beads-on-a-string preplatelets, which undergo fission into mature platelets. For the first time, we show that preplatelet maturation is a third, tightly regulated, critical process akin to cytokinesis that regulates platelet count. We show that deficiency in cytokine receptor-like factor 3 (CRLF3) in mice leads to an isolated and sustained 25% to 48% reduction in the platelet count without any effect on other blood cell lineages. We show that Crlf3−/− preplatelets have increased microtubule stability, possibly because of increased microtubule glutamylation via the interaction of CRLF3 with key members of the Hippo pathway. Using a mouse model of JAK2 V617F essential thrombocythemia, we show that a lack of CRLF3 leads to long-term lineage-specific normalization of the platelet count. We thereby postulate that targeting CRLF3 has therapeutic potential for treatment of thrombocythemia.
Introduction
Platelets are small (2-4 μm) anucleated blood cells, the main function of which is to form thrombi upon vessel injury. Thrombi can form inappropriately on atherothrombotic plaques, causing heart attacks or strokes. Platelets are produced by megakaryocytes (MKs), which derive from hematopoietic stem cells (HSCs). To release platelets, MKs produce long cytoskeletal processes (proplatelets), which extend into the circulation, where large fragments (preplatelets) are shed.1,2 Preplatelets undergo fission to form mature discoid platelets.3
Thrombocytopenia (platelet count <150 × 109/L) can be caused by lack of platelet production or peripheral consumption of platelets. Multiple genetic disorders affect platelet production, which can be broadly separated into 2 groups: disorders that affect MK differentiation (the first stage of platelet production) and disorders that affect proplatelet formation (the second stage). Unsurprisingly, mutations associated with the latter are often in genes associated with the actin-tubulin cytoskeleton, such as TUBB1,4MHY9,5FLNA,6ACTN1,7TPM4,8 and DIAPH1.9
Thrombocythemia (platelet count >450 × 109/L) resulting from acquired clonal mutations in HSCs is termed essential thrombocythemia (ET). The major mutations seen in ET affect the tyrosine kinase Janus kinase 2 (JAK2),10-13 the endoplasmic reticulum chaperone calreticulin,14,15 and the thrombopoietin (TPO) receptor MPL.16,17 Patients with ET typically have high survival rates, and the main complications are serious thrombotic events (affecting one-third of patients). The therapeutic management in patients with ET is primarily to prevent thrombotic events18 with agents that reduce platelet function (low-dose aspirin) and cytoreductive agents that reduce MK production (hydroxyurea and anagrelide).
Cytokine receptor-like factor 3 (CRLF3) is a poorly studied but widely expressed 488–amino acid protein encoded in chromosome 17q11.2 in a region that is deleted in neurofibromatosis type 1. CRLF3 is implicated in cell-cycle progression by overexpression in cell lines.19
We show for the first time that preplatelet fission to platelets is a critical rate-limiting step of platelet production (the third stage) and that CRLF3 plays a central role in this process by controlling microtubule stability, potentially through its interaction with Hippo pathway proteins. We also show that CRLF3 deficiency leads to isolated and sustained correction of platelet count in a mouse model of ET, showing its potential as a novel therapeutic target for ET.
Methods
Ethics
This research was regulated by the UK Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012 (project license 70/8406) and the district government of Lower Franconia (Bezirksregierung Unterfranken). Human blood samples were obtained from healthy volunteers under local ethics approval (HBREC.2018.13). Ethical approval for whole-genome sequencing was provided by the East of England Cambridge South national research ethics committee (13/EE/0325). Informed consent was obtained as per the Declaration of Helsinki.
Animals
Generation of Crlf3−/− (Crlf3tm1b(KOMP)Wtsi) mice was performed as previously reported.20-22 Mice were maintained on a C57Bl/6 background. Age- and sex-matched control animals were used in all experiments.
Complete blood counts
ETDA anticoagulated whole blood taken from the tail vein or inferior vena cava was run on an ABC blood counter (Woodley) or Vet abc Plus+ (Scil).
In vitro platelet assays
Platelet response to agonists,23 platelet spreading,24 expression of major platelet receptors,25 platelet survival,26 and cold-induced microtubule disassembly9 were performed as previously described. Thrombus formation assays were performed with heparin anticoagulated whole blood flowed into Vena8 Fluoro+ Biochips (Cellix) precoated with HORM Collagen (Takeda) as described.27 Five images per channel were obtained using an EVOS fl microscope and AMG camera and analyzed in ImageJ.
Platelet depletion
Mice were injected intraperitoneally with 0.6 µg/g of anti-CD42b (Emfret Analytics) in phosphate-buffered saline. Mice were bled for complete blood count from the tail vein at 0, 24, 48, 72, and 96 hours postinjection into EDTA-coated tubes (Microvette).
Splenectomy
Platelet and preplatelet counts were determined pre-/post-splenectomy by flow cytometry as described.28 Preplatelets were counted as GPV+/GPIIbIIIa+ events with larger forward/sideward scatter characteristics than platelets. Heparinized blood incubated with antibodies against activated GPIIbIIIa (M023-2) and CD62P (M130-1; both Emfret Analytics) eliminated preplatelets as microaggregates (supplemental Figure 4D, available on the Blood Web site).
Platelet imaging
Transmission and scanning electron microscopy, rapid Romanowsky staining, and confocal microscopy were performed as detailed in the data supplement. Two-photon intravital microscopy was performed as described.29
MK cultures
Mouse bone marrow (BM) cells were prepared, MKs cultured, and mature MKs purified as described.23,30 Differentiation and ploidy of cultured MKs were analyzed as described23 using propidium iodide (Sigma-Aldrich). Samples were acquired using a Beckman Coulter Cyan flow cytometer and analyzed using Kaluza Analysis (version 1.5a) software (Beckman Coulter). Details of in vitro proplatelet formation are provided in the data supplement.
iPSC MKs
Forward programming of tandem affinity purification (TAP)–tagged (data supplement) and untagged induced pluripotent stem cells (iPSCs) to iPSC MKs was performed as previously described.27 Proplatelet formation was carried out as described. For protein distribution, iPSC MKs attached to coverslips were fixed with 10% neutral buffered formalin (Sigma-Aldrich) and stained with antibodies against α-tubulin (T5168), FLAG (F1804; both Sigma-Aldrich), and 4′,6-diamidino-2-phenylindole (DAPI). Images were acquired using a Leica Sp5 inverted confocal microscope with a 63× immersion oil objective and Leica LAS 2.1 software and analyzed using ImageJ.
Structural solution of CRLF3
Purified murine CRLF3 protein (amino acids 174-442) was obtained using standard cloning, production, and purification methods. Crystallization was screened by the vapor diffusion method in 96-well sitting drop plates set up with a Nanodrop Screenmaker 96 + 8 (Innovadyne Technologies). Diffraction data were recorded at Diamond Light Source (Didcot, United Kingdom). The structure was determined by Hg-SAD using the AutoSol-wizard31 of the PHENIX suite32 with a data set collected with a wavelength of 1.006 Å from a crystal grown in 20% PEG 3350 and 0.2 M sodium formate (pH, 7.0) and soaked with 10 mM of thimerosal (Sigma-Aldrich) for 16 hours. The structure was refined to a resolution of 1.61 Å using Phenix.refine32 and by manual building in Coot.33 Full methodology is provided in the data supplement.
Genome-wide association studies
We performed a genetic association analysis of 3 loci (MOB1A, CRLF3, and STK38) to test for associations with 29 hematological parameters with imputed variants of minor allele frequency (MAF) >0.005% and INFO score >0.4. A significance threshold of 8.31 × 10−9 identifies associated variants located in each of the 3 genes by annotation with a variant effect predictor. Furthermore, we performed a multiple stepwise regression analysis to identify the number conditionally independent variants representing independent association signals in each locus.
Genetic variants in the human population
The primary data were obtained by whole-genome sequencing from whole-blood DNA from 13 037 individuals in the National Institute for Health Research BioResourceRare Diseases and 100 000 Genomes Project Pilot studies.34,35 Quality control, demographics, and variant calling are described in the report by Karczewski et al.34
Statistics
Sample sizes and statistical tests for each experiment are denoted in the figure legends (statistical testing was performed in Prism 8.0.1 [GraphPad Software]). A P value of <.05 was considered statistically significant (*P < .05, **P < .01, ***P < .005). Data are presented as means ± standard deviations.
Additional methods used in this study are provided in the data supplement.
Results
Crlf3 deficiency causes an isolated reduction in platelet count
Crlf3−/− mice were generated as part of a genome-wide screening program,20-22 leading to germ line deletion of Crlf3 exon 2. Despite Crlf3 being expressed in a large variety of tissues, including all hematopoietic lineages, Crlf3−/− animals show a sustained and isolated 25% to 48% reduction in platelet count (P < .005) compared with control (wild-type [WT]) animals (Figure 1A; supplemental Table 1), justifying further study of this mouse strain. Crlf3 messenger RNA was significantly reduced in cultured MKs from Crlf3−/− animals (Figure 1B), and CRLF3 protein was undetectable in both Crlf3−/− cultured MK (Figure 1C) and platelet lysates (data not shown).
![CRLF3 deficiency causes sustained and isolated reduction in platelet count. (A) Platelet counts of male (n = 5-23) and female (n = 5-14) young (12-20 weeks), middle aged (21-40 weeks), and old (>48 weeks) control (WT; blue) and Crlf3−/− (red) mice. (B) Expression of Crlf3 relative to Gapdh messenger RNA (mRNA) determined by quantitative reverse transcription polymerase chain reaction (qRT-PCR) of control (WT; blue) and Crlf3−/− (red) isolated from in vitro cultured MKs (n = 2). (C) Western blot of platelet lysates against CRLF3 (green) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; red; n = 2). (D) Platelet counts pre- (left; n = 15 WT and 14 Crlf3−/−) and 16 weeks post–BM transplantation (BMT; right) of control (WT; circles) and Crlf3−/− (squares) recipient mice that received either control (WT; blue) or Crlf3−/− (red) donor cells (n = 8 WT → WT; n = 7 all other groups). (E) Chimerism was estimated by expression of Crlf3 relative to Gapdh mRNA isolated from in vitro cultured MKs by qRT-PCR (n = 8 WT → WT; 7 all other groups). (F) Quantification of MKs in hematoxylin and eosin–stained sections of control (WT; blue) and Crlf3−/− (red) tibias (n = 6). (G) TPO concentration determined by enzyme-linked immunosorbent assay in control (WT; blue) and Crlf3−/− (red) plasma (n = 5 WT and 6 Crlf3−/−). (H) Percentage of CD41+ cells from control (WT; blue) and Crlf3−/− (red) in vitro MK cultures (n = 3). (I) Polyploidy of in vitro cultured control (WT; blue) and Crlf3−/− (red) MKs analyzed by flow cytometry (n = 5). (J) Mature in vitro cultured MKs were purified by bovine serum albumin gradient, seeded onto fibrinogen-coated coverslips and incubated at 37°C for 5 hours to induce proplatelet formation. Fixed samples were stained with CD41 (green) and DAPI (blue) and imaged by fluorescence microscopy. Images are representative of Crlf3−/− and control (WT) proplatelet-forming MKs. Scale bars are 50 μm. Proplatelet morphology of control (WT; blue) and Crlf3−/− (red) MKs was assessed by blindly quantifying the number of protrusions per proplatelet-forming MK and number of branches per protrusion (n = 29 WT and 31 Crlf3−/−). (K) In vitro cultured MKs were seeded onto fibrinogen-coated coverslips and incubated at 37°C for 3 or 5 hours to induce proplatelet formation. After confocal microscopy, percentage of proplatelet forming MKs was determined for control (WT; blue) and Crlf3−/− (red; n = 3). At least 460 MKs were counted in each condition. (L) Control (WT; blue) and Crlf3−/− (red) animals were injected with phosphate-buffered saline (PBS; circles) or anti-CD42b (0.6 μg/g body weight; squares) and platelet counts determined by automated hemocytometer 0, 24, 48, 72, and 96 hours postinjection (n = 4 Crlf3−/− plus CD42b antibody [Ab]; n = 3 all other groups). (M) Control (WT; blue) and Crlf3−/− (red) mice were injected with 1 mg of NHS-biotin, and percentage of CD41+/Ter119−/streptavidin+ platelets was determined by flow cytometry at 24, 48, 72, 96, and 168 hours postinjection. Percentage of streptavidin+ platelets at 24 hours represents 100% biotin-bound platelets (n = 5). Data represent means ± standard deviations. Unpaired 2-tailed Student t test (F-H,J) with correction for multiple comparisons using the Holm-Sidak method (A), 1-way analysis of variance (ANOVA) (D,E), or 2-way ANOVA (I,K-M) with correction for multiple comparisons using the Holm-Sidak method. *P < .05, **P < .01, ***P < .005. HPF, high-powered field; ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/14/10.1182_blood.2021013113/8/m_bloodbld2021013113f1.png?Expires=1765884566&Signature=fKvTCGd~CeTJdHfJFzzVvJh~26TG2fID3Qo-DKTvDRtohWueJdUJbAfDl2innT0PFWP67qHnd6l1IwanIt8KUSTotdaHtxAlmrNnO8dXxiY6lujxGL8B3AD73ai~sRa871HDgZ0NqqwKjfTYuScFVzZCP7tKxeuVJMWl3MK1IoHc19wVHlhct98DDxuF0aHLs8CYvkFgjSSeezWbuXIqKx94JKcbCaNvVvNuVJd7GkGZ0U9d1Cy1-c9FttKiKXQlQIvojBDnh9O1SS3yLS5gLJc9NwlmIsw6SX3Sm5fO-Zlf866vgBg6XdORyq7r2~4DfqgRnvoLRDeqJU2dVmBQFg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CRLF3 deficiency causes sustained and isolated reduction in platelet count. (A) Platelet counts of male (n = 5-23) and female (n = 5-14) young (12-20 weeks), middle aged (21-40 weeks), and old (>48 weeks) control (WT; blue) and Crlf3−/− (red) mice. (B) Expression of Crlf3 relative to Gapdh messenger RNA (mRNA) determined by quantitative reverse transcription polymerase chain reaction (qRT-PCR) of control (WT; blue) and Crlf3−/− (red) isolated from in vitro cultured MKs (n = 2). (C) Western blot of platelet lysates against CRLF3 (green) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; red; n = 2). (D) Platelet counts pre- (left; n = 15 WT and 14 Crlf3−/−) and 16 weeks post–BM transplantation (BMT; right) of control (WT; circles) and Crlf3−/− (squares) recipient mice that received either control (WT; blue) or Crlf3−/− (red) donor cells (n = 8 WT → WT; n = 7 all other groups). (E) Chimerism was estimated by expression of Crlf3 relative to Gapdh mRNA isolated from in vitro cultured MKs by qRT-PCR (n = 8 WT → WT; 7 all other groups). (F) Quantification of MKs in hematoxylin and eosin–stained sections of control (WT; blue) and Crlf3−/− (red) tibias (n = 6). (G) TPO concentration determined by enzyme-linked immunosorbent assay in control (WT; blue) and Crlf3−/− (red) plasma (n = 5 WT and 6 Crlf3−/−). (H) Percentage of CD41+ cells from control (WT; blue) and Crlf3−/− (red) in vitro MK cultures (n = 3). (I) Polyploidy of in vitro cultured control (WT; blue) and Crlf3−/− (red) MKs analyzed by flow cytometry (n = 5). (J) Mature in vitro cultured MKs were purified by bovine serum albumin gradient, seeded onto fibrinogen-coated coverslips and incubated at 37°C for 5 hours to induce proplatelet formation. Fixed samples were stained with CD41 (green) and DAPI (blue) and imaged by fluorescence microscopy. Images are representative of Crlf3−/− and control (WT) proplatelet-forming MKs. Scale bars are 50 μm. Proplatelet morphology of control (WT; blue) and Crlf3−/− (red) MKs was assessed by blindly quantifying the number of protrusions per proplatelet-forming MK and number of branches per protrusion (n = 29 WT and 31 Crlf3−/−). (K) In vitro cultured MKs were seeded onto fibrinogen-coated coverslips and incubated at 37°C for 3 or 5 hours to induce proplatelet formation. After confocal microscopy, percentage of proplatelet forming MKs was determined for control (WT; blue) and Crlf3−/− (red; n = 3). At least 460 MKs were counted in each condition. (L) Control (WT; blue) and Crlf3−/− (red) animals were injected with phosphate-buffered saline (PBS; circles) or anti-CD42b (0.6 μg/g body weight; squares) and platelet counts determined by automated hemocytometer 0, 24, 48, 72, and 96 hours postinjection (n = 4 Crlf3−/− plus CD42b antibody [Ab]; n = 3 all other groups). (M) Control (WT; blue) and Crlf3−/− (red) mice were injected with 1 mg of NHS-biotin, and percentage of CD41+/Ter119−/streptavidin+ platelets was determined by flow cytometry at 24, 48, 72, 96, and 168 hours postinjection. Percentage of streptavidin+ platelets at 24 hours represents 100% biotin-bound platelets (n = 5). Data represent means ± standard deviations. Unpaired 2-tailed Student t test (F-H,J) with correction for multiple comparisons using the Holm-Sidak method (A), 1-way analysis of variance (ANOVA) (D,E), or 2-way ANOVA (I,K-M) with correction for multiple comparisons using the Holm-Sidak method. *P < .05, **P < .01, ***P < .005. HPF, high-powered field; ns, not significant.
CRLF3 deficiency causes sustained and isolated reduction in platelet count. (A) Platelet counts of male (n = 5-23) and female (n = 5-14) young (12-20 weeks), middle aged (21-40 weeks), and old (>48 weeks) control (WT; blue) and Crlf3−/− (red) mice. (B) Expression of Crlf3 relative to Gapdh messenger RNA (mRNA) determined by quantitative reverse transcription polymerase chain reaction (qRT-PCR) of control (WT; blue) and Crlf3−/− (red) isolated from in vitro cultured MKs (n = 2). (C) Western blot of platelet lysates against CRLF3 (green) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; red; n = 2). (D) Platelet counts pre- (left; n = 15 WT and 14 Crlf3−/−) and 16 weeks post–BM transplantation (BMT; right) of control (WT; circles) and Crlf3−/− (squares) recipient mice that received either control (WT; blue) or Crlf3−/− (red) donor cells (n = 8 WT → WT; n = 7 all other groups). (E) Chimerism was estimated by expression of Crlf3 relative to Gapdh mRNA isolated from in vitro cultured MKs by qRT-PCR (n = 8 WT → WT; 7 all other groups). (F) Quantification of MKs in hematoxylin and eosin–stained sections of control (WT; blue) and Crlf3−/− (red) tibias (n = 6). (G) TPO concentration determined by enzyme-linked immunosorbent assay in control (WT; blue) and Crlf3−/− (red) plasma (n = 5 WT and 6 Crlf3−/−). (H) Percentage of CD41+ cells from control (WT; blue) and Crlf3−/− (red) in vitro MK cultures (n = 3). (I) Polyploidy of in vitro cultured control (WT; blue) and Crlf3−/− (red) MKs analyzed by flow cytometry (n = 5). (J) Mature in vitro cultured MKs were purified by bovine serum albumin gradient, seeded onto fibrinogen-coated coverslips and incubated at 37°C for 5 hours to induce proplatelet formation. Fixed samples were stained with CD41 (green) and DAPI (blue) and imaged by fluorescence microscopy. Images are representative of Crlf3−/− and control (WT) proplatelet-forming MKs. Scale bars are 50 μm. Proplatelet morphology of control (WT; blue) and Crlf3−/− (red) MKs was assessed by blindly quantifying the number of protrusions per proplatelet-forming MK and number of branches per protrusion (n = 29 WT and 31 Crlf3−/−). (K) In vitro cultured MKs were seeded onto fibrinogen-coated coverslips and incubated at 37°C for 3 or 5 hours to induce proplatelet formation. After confocal microscopy, percentage of proplatelet forming MKs was determined for control (WT; blue) and Crlf3−/− (red; n = 3). At least 460 MKs were counted in each condition. (L) Control (WT; blue) and Crlf3−/− (red) animals were injected with phosphate-buffered saline (PBS; circles) or anti-CD42b (0.6 μg/g body weight; squares) and platelet counts determined by automated hemocytometer 0, 24, 48, 72, and 96 hours postinjection (n = 4 Crlf3−/− plus CD42b antibody [Ab]; n = 3 all other groups). (M) Control (WT; blue) and Crlf3−/− (red) mice were injected with 1 mg of NHS-biotin, and percentage of CD41+/Ter119−/streptavidin+ platelets was determined by flow cytometry at 24, 48, 72, 96, and 168 hours postinjection. Percentage of streptavidin+ platelets at 24 hours represents 100% biotin-bound platelets (n = 5). Data represent means ± standard deviations. Unpaired 2-tailed Student t test (F-H,J) with correction for multiple comparisons using the Holm-Sidak method (A), 1-way analysis of variance (ANOVA) (D,E), or 2-way ANOVA (I,K-M) with correction for multiple comparisons using the Holm-Sidak method. *P < .05, **P < .01, ***P < .005. HPF, high-powered field; ns, not significant.
To assess whether the thrombocytopenia in Crlf3−/− animals was driven by factors intrinsic to the hematopoietic compartment, we performed BMTs. Control or Crlf3−/− BM cells were transplanted into irradiated control or Crlf3−/− recipient mice. Where donor and recipient genotypes were matched, the differences in platelet count determined pre-BMT remained true (Figure 1D). However, when WT recipients received Crlf3−/− BM, the platelet count post-BMT decreased to levels comparable to those in Crlf3−/− recipients that received Crlf3−/− BM (P = .9965). In contrast, when Crlf3−/− recipients received WT BM, the platelet count increased, reaching levels comparable to those in WT recipients that received WT BM (P = .9650). We confirmed that the posttransplantation platelet count correlated with Crlf3 expression in cultured MKs derived from recipient BM samples (Figure 1E).
Next, we sought to clarify whether the thrombocytopenia was caused by decreased platelet production and/or increased platelet clearance. MK differentiation was preserved; Crlf3−/− mice had increased BM MKs (MKs per field, 12.65 ± 1.03 for Crlf3−/− vs 8.90 ± 2.51 for WT; P = .0069; Figure 1F; supplemental Figure 1), TPO concentrations were only marginally increased in Crlf3−/− mice (253 ± 136 vs 201 ± 56 pg/mL; P = .4500; Figure 1G), Crlf3−/− BM samples cultured in a suboptimal concentration of TPO showed a higher percentage of CD41+ cells after 5 days compared with controls (55.4% ± 7.1% vs 29.7% ± 2.5%; P = .0042; Figure 1H), and ploidy of cultured MKs was unchanged (Figure 1I). Proplatelet formation was morphologically similar between Crlf3−/− and control cultured MKs (Figure 1J), showing similar numbers of protrusions (P = .2989; Figure 1J, left panel) and branching (P = .9226; Figure 1J, right panel). However, proplatelet dynamics appeared altered between cultured MKs from Crlf3−/− and control animals. A greater proportion of Crlf3−/− MKs formed proplatelets 3 hours postseeding onto fibrinogen (45% ± 8% vs 31% ± 13%; P = .1038; Figure 1K; supplemental Figure 2), whereas at 5 hours, the trend was reversed (26% ± 2% vs 52% ± 10%; P = .0164). We presume the data at 5 hours reflect proplatelet-forming MKs seen at 3 hours in the Crlf3−/− sample having fragmented into platelets at 5 hours, which is supported by the reduced density of Crlf3−/− MKs at 5 hours (supplemental Figure 2). Using in vivo 2-photon intravital microscopy, we confirmed Crlf3−/− MKs formed long proplatelet protrusions into BM sinusoids, which appeared to be no different from those seen in control animals (supplemental Videos 1 and 2). Finally, we assessed platelet recovery after platelet depletion. Platelet counts in depleted Crlf3−/− and control animals recovered at an indistinguishable rate, with platelet counts reaching their respective values before depletion in 96 hours (Figure 1L). These data confirm that Crlf3−/− MKs differentiate normally and can produce platelets at least at a normal rate. The slight increase in MKs would imply a compensatory mechanism to increased platelet consumption.
We next considered whether the reduced platelet count was due to abnormal platelet function and/or clearance. Crlf3−/− mice do not display any overt bleeding phenotype. We carried out a tail bleeding assay, and 1 Crlf3−/− animal did show increased blood loss in the early time point compared with controls and its Crlf3−/− littermate upon tail transection (supplemental Figure 3A). We confirmed there were no gross differences in any main platelet functions, namely adhesion, spreading, activation, or thrombus formation. The expression of key platelet surface receptors/integrins was similar (P > .05 for all tested; supplemental Figure 3B). Platelet activation measured as fibrinogen binding and P-selectin surface expression by flow cytometry was comparable in response to different agonists (P > .05 for all agonists at all doses; supplemental Figure 3C). Platelet spreading onto fibrinogen was also not different (P = .7717; supplemental Figure 3D). Thrombus formation of whole blood flowed at arterial shear rates over a collagen-coated surface was equally efficient (P = .9809; supplemental Figure 3E). Finally, we determined platelet lifespan by flow cytometry. We found that the gradual decrease in labeled platelets was identical in both control and Crlf3−/− animals, suggesting that platelet lifespan is unaffected (Figure 1M).
Crlf3 deficiency leads to ineffective thrombopoiesis
Preplatelets released into the BM sinusoids resemble proplatelet shafts, barbell platelets, or giant platelets.1,2 Preplatelets were rarely seen on the blood smears of control mice (supplemental Figure 4A) but were easily identified on Crlf3−/− samples. Remarkably, some of these were several hundred microns in length, with the classical beads-on-a-string appearance, which has been reported before in culture but not in the peripheral circulation (Figure 2A; supplemental Figure 4A). We confirmed that these structures were preplatelets using immunofluorescence staining for specific platelet cell surface markers (CD41) and proteins contained in platelet α-granules, including von Willebrand factor (Figure 2B), and using scanning (Figure 2C; supplemental Figure 4B) and transmission (Figure 2D; supplemental Figure 4C) electron microscopy. We hypothesized that lack of CRLF3 impairs preplatelet fission and that a proportion of these circulating hyperstable preplatelets are removed from the peripheral circulation (primarily in the spleen) before they have the chance to mature into platelets. This would decrease the number of new platelets produced by each MK (ineffective thrombopoiesis). We hypothesized that splenectomy would allow the preplatelets to circulate longer, allowing them to undergo fission and correct the platelet count. We measured circulating platelet and preplatelet counts by flow cytometry pre- and postsplenectomy by a published method3 (Figure 2E). Spleen size and weight as well as histology were comparable between Crlf3−/− and control animals (supplemental Figure 4E). Control animals showed a slight increase in the platelet count as expected postsplenectomy (Figure 2F). The platelet count in splenectomized Crlf3−/− animals also increased postsurgery but crucially reached the same level as that seen in the control animals (P = .4344) despite being 38% lower before splenectomy (P < .0001). Preplatelets were more abundant in the Crlf3−/− animals presplenectomy (P = .0010; Figure 2G). Postsplenectomy, circulating preplatelets increased marginally in control animals but decreased in Crlf3−/− animals to levels like those seen in the controls (P = .2524; Figure 2G). We postulate that splenectomy allows Crlf3−/− preplatelets to circulate for long enough to mature into platelets (switching from ineffective to effective thrombopoiesis), thereby improving the number of platelets produced per MK. In keeping with this, postsplenectomy, MK numbers in the BM of Crlf3−/− animals reduced toward levels seen in control animals (Figure 2H).
![CRLF3 deficiency causes ineffective thrombopoiesis. (A) Romanovsky-stained blood smear from Crlf3−/− mouse whole blood taken at 100× magnification under light microscopy. (B) Crlf3−/− blood smear stained with CD41 (green) and von Willebrand factor (VWF; red) and imaged by confocal microscopy. (C,D) Washed Crlf3−/− platelets fixed and prepared for scanning (C) or transmission electron microscopy (D). Scale bars are 5 μm (B,C) and 2 μm (D). (E-G) Example flow cytometric plots (E) to determine platelet (GPV+/GPIIbIIIa+ events) (F) and preplatelet (GPV+/GPIIbIIIa+ events with larger forward scatter [FSC]/side scatter [SSC] than mature platelets) (G) counts from control (WT; blue) and Crlf3−/− (red) splenectomized mice (n = 4 Crlf3−/− postsplenectomy; n = 5 all other groups). (H) Quantification of MKs in hematoxylin and eosin–stained sections of control (WT; blue) and Crlf3−/− (red) tibia of nonsplenectomized animals (left) or 21 postsplenectomy (right; n = 6 non-splenectomized and 2 splenectomized). Data represent means ± standard deviations (except splenectomized mice in panel H, where data represent means). Two-way analysis of variance with correction for multiple comparisons using the Holm-Sidak method (F,G) and unpaired 2-tailed Student t test (H). **P < .01, ***P < .005. FITC, fluorescein isothiocyanate; HPF, high-powered field; ns, not significant; PE, phycoerythrin.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/14/10.1182_blood.2021013113/8/m_bloodbld2021013113f2.png?Expires=1765884566&Signature=qV7cW2BCDFJ5AAnXtvbjYH9MzxXY8ePMVMc2KPKtuBOYxoEX8GTMel~MBp7B9qwIpAXL15T5uXAXq-sBcwMVx4YCn5jzI1oXr61clSxOpxpqAdfn2Ee9vWoKA3ZqSo4uaBp8RrhxF~CNudc8cGjGoWLI4MlJSO3xIM-1ei-jYF3XGiF1PtkNG7pnNj~hqfpPj4Qd4pNwH4KRUg8SDwK2MiTOOQsTmzVjucMxpTB26NrFfa33UX1lhpxUUZcs~sUlMgWPZXN3NOPaUrzPJrNPOBvacgk0u0puH1OoLpJ9lBKtetGMz~76aBUZgIgYCUJP-9r715ytSZXycNwNZuSTkg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CRLF3 deficiency causes ineffective thrombopoiesis. (A) Romanovsky-stained blood smear from Crlf3−/− mouse whole blood taken at 100× magnification under light microscopy. (B) Crlf3−/− blood smear stained with CD41 (green) and von Willebrand factor (VWF; red) and imaged by confocal microscopy. (C,D) Washed Crlf3−/− platelets fixed and prepared for scanning (C) or transmission electron microscopy (D). Scale bars are 5 μm (B,C) and 2 μm (D). (E-G) Example flow cytometric plots (E) to determine platelet (GPV+/GPIIbIIIa+ events) (F) and preplatelet (GPV+/GPIIbIIIa+ events with larger forward scatter [FSC]/side scatter [SSC] than mature platelets) (G) counts from control (WT; blue) and Crlf3−/− (red) splenectomized mice (n = 4 Crlf3−/− postsplenectomy; n = 5 all other groups). (H) Quantification of MKs in hematoxylin and eosin–stained sections of control (WT; blue) and Crlf3−/− (red) tibia of nonsplenectomized animals (left) or 21 postsplenectomy (right; n = 6 non-splenectomized and 2 splenectomized). Data represent means ± standard deviations (except splenectomized mice in panel H, where data represent means). Two-way analysis of variance with correction for multiple comparisons using the Holm-Sidak method (F,G) and unpaired 2-tailed Student t test (H). **P < .01, ***P < .005. FITC, fluorescein isothiocyanate; HPF, high-powered field; ns, not significant; PE, phycoerythrin.
CRLF3 deficiency causes ineffective thrombopoiesis. (A) Romanovsky-stained blood smear from Crlf3−/− mouse whole blood taken at 100× magnification under light microscopy. (B) Crlf3−/− blood smear stained with CD41 (green) and von Willebrand factor (VWF; red) and imaged by confocal microscopy. (C,D) Washed Crlf3−/− platelets fixed and prepared for scanning (C) or transmission electron microscopy (D). Scale bars are 5 μm (B,C) and 2 μm (D). (E-G) Example flow cytometric plots (E) to determine platelet (GPV+/GPIIbIIIa+ events) (F) and preplatelet (GPV+/GPIIbIIIa+ events with larger forward scatter [FSC]/side scatter [SSC] than mature platelets) (G) counts from control (WT; blue) and Crlf3−/− (red) splenectomized mice (n = 4 Crlf3−/− postsplenectomy; n = 5 all other groups). (H) Quantification of MKs in hematoxylin and eosin–stained sections of control (WT; blue) and Crlf3−/− (red) tibia of nonsplenectomized animals (left) or 21 postsplenectomy (right; n = 6 non-splenectomized and 2 splenectomized). Data represent means ± standard deviations (except splenectomized mice in panel H, where data represent means). Two-way analysis of variance with correction for multiple comparisons using the Holm-Sidak method (F,G) and unpaired 2-tailed Student t test (H). **P < .01, ***P < .005. FITC, fluorescein isothiocyanate; HPF, high-powered field; ns, not significant; PE, phycoerythrin.
Crlf3−/− MKs contain hyperstable polyglutamylated microtubules
Platelet genesis is driven by microtubule assembly and reorganization. Proplatelet formation and preplatelet release rely on microtubule formation in the proplatelet shaft, whereas preplatelet maturation into mature platelets requires tubulin bundle twisting, followed by disassembly and severing. Tubulin staining in control platelets and a majority of the mature Crlf3−/− platelets showed the classical peripheral coil (Figure 3A-D). However, some Crlf3−/− platelets had disorganized tubulin, particularly in preplatelets (Figure 3C-D). Most control platelets fully disassembled their microtubule coil upon cooling to 4°C, whereas a significantly larger proportion of Crlf3−/− platelets retained at least partially some of the marginal band (41% ± 4% vs 14% ± 1%; P = .0003; Figure 3E-I). Microtubule stability is influenced by posttranslational modifications (PTMs), such as tyrosination, acetylation, or glutamylation.26-28 We saw no difference in the total tubulin content (P = .1978), tubulin tyrosination (P = .7218), or tubulin acetylation (P = .1312) of cultured Crlf3−/− MKs, normalized to total tubulin content (Figure 3J; supplemental Figure 5A-C). However, polyglutamylated tubulin content appeared increased in cultured Crlf3−/− MKs, although not reaching significance (1.85-fold increase; P = .0713; Figure 3J). We therefore probed MK samples with an alternative antibody against polyglutamylated tubulin and showed a similar small increase in polyglutamylated tubulin content (1.66-fold; P = .0050; supplemental Figure 5D). Platelet tubulin content and modified tubulins were also analyzed but failed to reveal any significant differences; however, there was a trend toward increased tyrosinated tubulin in platelets (1.47-fold increase; P = .0639; Figure 3K; supplemental Figure 5A-C). We postulate that polyglutamylated tubulin was unchanged in platelets, because those would be most likely arising from MKs with the lowest level of glutamylation, allowing for prompt proplatelet maturation. Using immunofluorescence, we showed bundles of glutamylated tubulin leading toward the proplatelet shafts in some Crlf3−/− MKs (Figure 3L), but this was not the case in all cells analyzed (supplemental Figure 5E).
![CRLF3 deficiency causes microtubule hyperstability. (A-H) Washed platelets maintained at 37°C (control [WT]) (A,B) (Crlf3−/−) (C,D) or stored at 4°C for 3 hours (control [WT]) (E,F) (Crlf3−/−) (G,H) adhered to poly-L-lysine–coated coverslips and stained for α-tubulin (green) and F-actin (red). Scale bars are 20 μm (A,C,E,G) and 5 μm (B,D,F,H). (I) Platelets retaining microtubule structures after incubation at 4°C were determined by manual counting of images for control (WT; blue) and Crlf3−/− (red) mice (n = 3). (J-K) Representative western blots of in vitro cultured MK (J) or platelet (K) lysates against tyrosine α-tubulin, α-tubulin, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; left); acetylated α-tubulin, α-tubulin, and GAPDH (middle); or glutamylated α-tubulin (AG-20B-0020_upper band), α-tubulin, and GAPDH (right) for control (WT; blue) and Crlf3−/− (red) samples. The quantification of glutamylated α-tubulin/total tubulin in in vitro cultured MKs (bar graphs on right) was carried out on 8 control and 8 Crlf3−/− samples with 2 technical replicates. Where α-tubulin and GAPDH panels are the same for multiple tubulin modifications, membranes were stripped and reprobed between antibodies against specific tubulin modifications before finally being stripped and reprobed for α-tubulin and GAPDH. (L) In vitro cultured MKs were seeded onto fibrinogen-coated coverslips and incubated at 37°C for 5 hours to induce proplatelet formation. Samples were fixed, stained for polyglutamylated α-tubulin (AG-20B-0020), and imaged by fluorescence microscopy. Images are representative for Crlf3−/− and control (WT) proplatelet-forming MKs. Scale bars are 50 μm. Data represent means ± standard deviations. Unpaired 2-tailed Student (I,K) or Welch (J) t test. ***P < .005.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/14/10.1182_blood.2021013113/8/m_bloodbld2021013113f3.png?Expires=1765884566&Signature=x2qZL4lv3xsZIkUZuXCBsLDt682Gono9S1cch8IWXCO3Kchpz2Ynm1EyiZTXGAi1Xr5tXwYGn-YHKZRSlfHnFbhlIaTy~6ALlJnajQtC4oZ-EAQODUNkGusg8OOdVtPf0XRWdN3CtYiwioB-jxu-kPfwnQgwdMqAWclDv2W4JU~3Roy3TA6o9qNz-4QQ0jOG6DKoDsHTHg0FBk3A~0EV-E4Re3l2vbCtrHh7VurCANXZGK~5BR4IzyU3pwzrUOl~7ddKZMP3Nr~YybUeXQp4TT5tPfXJRSZx0EKsCnACXZcs0H~TSyzNV9mHA2FJBR2sg-7nyl2hpkiousILvanPNA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CRLF3 deficiency causes microtubule hyperstability. (A-H) Washed platelets maintained at 37°C (control [WT]) (A,B) (Crlf3−/−) (C,D) or stored at 4°C for 3 hours (control [WT]) (E,F) (Crlf3−/−) (G,H) adhered to poly-L-lysine–coated coverslips and stained for α-tubulin (green) and F-actin (red). Scale bars are 20 μm (A,C,E,G) and 5 μm (B,D,F,H). (I) Platelets retaining microtubule structures after incubation at 4°C were determined by manual counting of images for control (WT; blue) and Crlf3−/− (red) mice (n = 3). (J-K) Representative western blots of in vitro cultured MK (J) or platelet (K) lysates against tyrosine α-tubulin, α-tubulin, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; left); acetylated α-tubulin, α-tubulin, and GAPDH (middle); or glutamylated α-tubulin (AG-20B-0020_upper band), α-tubulin, and GAPDH (right) for control (WT; blue) and Crlf3−/− (red) samples. The quantification of glutamylated α-tubulin/total tubulin in in vitro cultured MKs (bar graphs on right) was carried out on 8 control and 8 Crlf3−/− samples with 2 technical replicates. Where α-tubulin and GAPDH panels are the same for multiple tubulin modifications, membranes were stripped and reprobed between antibodies against specific tubulin modifications before finally being stripped and reprobed for α-tubulin and GAPDH. (L) In vitro cultured MKs were seeded onto fibrinogen-coated coverslips and incubated at 37°C for 5 hours to induce proplatelet formation. Samples were fixed, stained for polyglutamylated α-tubulin (AG-20B-0020), and imaged by fluorescence microscopy. Images are representative for Crlf3−/− and control (WT) proplatelet-forming MKs. Scale bars are 50 μm. Data represent means ± standard deviations. Unpaired 2-tailed Student (I,K) or Welch (J) t test. ***P < .005.
CRLF3 deficiency causes microtubule hyperstability. (A-H) Washed platelets maintained at 37°C (control [WT]) (A,B) (Crlf3−/−) (C,D) or stored at 4°C for 3 hours (control [WT]) (E,F) (Crlf3−/−) (G,H) adhered to poly-L-lysine–coated coverslips and stained for α-tubulin (green) and F-actin (red). Scale bars are 20 μm (A,C,E,G) and 5 μm (B,D,F,H). (I) Platelets retaining microtubule structures after incubation at 4°C were determined by manual counting of images for control (WT; blue) and Crlf3−/− (red) mice (n = 3). (J-K) Representative western blots of in vitro cultured MK (J) or platelet (K) lysates against tyrosine α-tubulin, α-tubulin, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; left); acetylated α-tubulin, α-tubulin, and GAPDH (middle); or glutamylated α-tubulin (AG-20B-0020_upper band), α-tubulin, and GAPDH (right) for control (WT; blue) and Crlf3−/− (red) samples. The quantification of glutamylated α-tubulin/total tubulin in in vitro cultured MKs (bar graphs on right) was carried out on 8 control and 8 Crlf3−/− samples with 2 technical replicates. Where α-tubulin and GAPDH panels are the same for multiple tubulin modifications, membranes were stripped and reprobed between antibodies against specific tubulin modifications before finally being stripped and reprobed for α-tubulin and GAPDH. (L) In vitro cultured MKs were seeded onto fibrinogen-coated coverslips and incubated at 37°C for 5 hours to induce proplatelet formation. Samples were fixed, stained for polyglutamylated α-tubulin (AG-20B-0020), and imaged by fluorescence microscopy. Images are representative for Crlf3−/− and control (WT) proplatelet-forming MKs. Scale bars are 50 μm. Data represent means ± standard deviations. Unpaired 2-tailed Student (I,K) or Welch (J) t test. ***P < .005.
CRLF3 interacts with STK38, and its absence leads to increased MOB1 phosphorylation
To gain a mechanistic understanding the role of CRLF3 in tubulin glutamylation, we switched over to a human cellular system. First, we sought to identify the CRLF3 cellular localization and protein partners in relevant cells (ie, platelets and MKs). Human platelet lysates were subfractionated by sucrose gradient centrifugation.36 Western blot analysis clearly showed enrichment of CRLF3 in the subfractions containing cytoskeletal proteins, particularly α-tubulin (Figure 4A). To refine the localization of CRLF3 and perform pull-down experiments, we used a human iPSC–based system. We inserted a TAP tag37 at the 3′ end of the endogenous CRLF3 gene in iPSCs. Tagged and untagged control iPSCs were differentiated into highly pure populations of MKs (Figure 4B) by forward programming.27 The expression of CRLF3 TAP was confirmed in both tagged iPSCs and their MK progeny (iPSC MKs; Figure 4C). In non–proplatelet-forming iPSC MKs, CRLF3 TAP showed a diffuse mainly cytoplasmic pattern. By contrast, in proplatelet-forming iPSC MKs, CRLF3 TAP appears to redistribute to the plasma membrane (Figure 4D). We went on to perform mass spectrometry on anti-FLAG immunoprecipitation samples from CRLF3 TAP and control iPSC MKs, and several candidate interacting proteins were identified (supplemental Table 2). One candidate interactor was STK38, a member of a group of NDR kinases known to interact with MOB1.38 MOB1 is a key member of the Hippo pathway39 and a protein that has been shown to influence tubulin stability through PTMs.40 MOB1 has previously been shown to interact with CRLF3 in HEK cells treated with okadaic acid.41,42 We performed anti-FLAG immunoprecipitation followed by western blotting on okadaic acid–treated CRLF3-tagged iPSC MKs and confirmed the interaction between CRLF3 and STK38 (Figure 4E). We could not show evidence of an interaction between CRLF3 and MOB1 in either the forward or reverse pull downs (Figure 4E); however, in anti-MOB1 immunoprecipitated control iPSC MKs, we confirmed the interaction between MOB1 and STK38 (Figure 4F). We did not see a difference in MOB1 localization (Figure 4G) or total quantity of MOB1 (P = .3337; Figure 4H) in Crlf3−/− MKs. However, we saw increased phosphorylation of MOB1 (>2.5-fold; P = .0286; Figure 4H; supplemental Figure 6). Because we had established an interaction between CRLF3 and STK38 in MKs, we looked at the influence of CRLF3 on STK38 protein. We saw a small (∼1.5-fold) nonsignificant increase in total quantity of STK38 (P = .1112; Figure 4H) in Crlf3−/− MKs, but STK38 phosphorylation was unchanged (P = .4398).
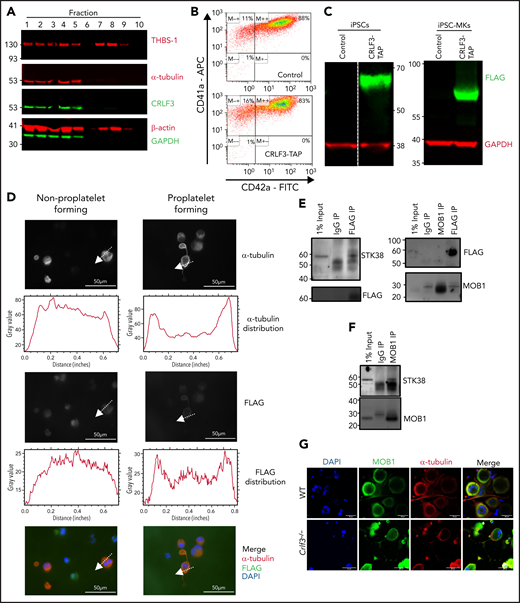
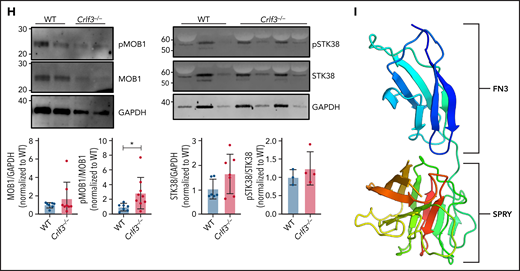
CRLF3 interacts with the Hippo pathway. (A) Western blot of sucrose gradient centrifugation fractionated human platelets probed with antibodies against α-tubulin, β-actin, thrombospondin (THBS-1), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and CRLF3. Fractions 1 to 5 represent cytoskeletal proteins (enriched for α-tubulin and β-actin), whereas fractions 7 and 8 represent granular proteins (enriched for THBS-1). (B) Representative flow cytometric plots of CRLF3 TAP-tagged and control forward programmed iPSC MKs stained with CD41a and CD42a. (C) Western blot of CRLF3 TAP-tagged and control iPSCs and iPSC MKs probed with antibodies against FLAG (green) and GAPDH (red). (D) CRLF3 TAP-tagged iPSC MKs were seeded onto fibrinogen-coated coverslips and incubated at 37°C for 24 hours to induce proplatelet formation. Samples were fixed, stained with α-tubulin (red), FLAG (green), and DAPI (blue), and imaged by fluorescence microscopy. Subcellular distribution of α-tubulin and FLAG staining in round and proplatelet-forming CRLF3 TAP iPSC MKs was determined across a section of the MKs along the indicated arrow using ImageJ. Scale bars are 50 μm. (E-F) CRLF3 TAP (E) and control (F) iPSC MKs were lysed and immunoprecipitated (IP) with antibodies against FLAG, MOB1, and immunoglobulin G (IgG). Precipitated lysates were then probed for STK38, MOB1, and FLAG by western blot. (G) In vitro cultured MKs were seeded onto fibrinogen-coated coverslips and incubated at 37°C for 5 hours to induce proplatelet formation. Samples were fixed, stained for MOB1 (green), α-tubulin (red), and DAPI (blue), and imaged by fluorescence microscopy. Images are representative for Crlf3−/− and control (WT) proplatelet-forming MKs. (H) Western blot of in vitro cultured MKs probed with antibodies against pMOB1, MOB1, and GAPDH (left panel; n = 8 MOB1/GAPDH and 4 pMOB1/GAPDH) and pSTK38, STK38 and GAPDH (right panel; n = 3 STK38/GAPDH, 3 Crlf3−/−, and 4 WT pSTK38/GAPDH). (I) Three-dimensional structure of CRLF3 (residue 174 to end) solved by experimental phasing. Domains are labeled. Molecular graphics prepared using PyMOL. Data represent means ± standard deviations. Unpaired 2-tailed Student t test. *P < .05. FITC, fluorescein isothiocyanate; FN3, fibronectin type 3.
CRLF3 interacts with the Hippo pathway. (A) Western blot of sucrose gradient centrifugation fractionated human platelets probed with antibodies against α-tubulin, β-actin, thrombospondin (THBS-1), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and CRLF3. Fractions 1 to 5 represent cytoskeletal proteins (enriched for α-tubulin and β-actin), whereas fractions 7 and 8 represent granular proteins (enriched for THBS-1). (B) Representative flow cytometric plots of CRLF3 TAP-tagged and control forward programmed iPSC MKs stained with CD41a and CD42a. (C) Western blot of CRLF3 TAP-tagged and control iPSCs and iPSC MKs probed with antibodies against FLAG (green) and GAPDH (red). (D) CRLF3 TAP-tagged iPSC MKs were seeded onto fibrinogen-coated coverslips and incubated at 37°C for 24 hours to induce proplatelet formation. Samples were fixed, stained with α-tubulin (red), FLAG (green), and DAPI (blue), and imaged by fluorescence microscopy. Subcellular distribution of α-tubulin and FLAG staining in round and proplatelet-forming CRLF3 TAP iPSC MKs was determined across a section of the MKs along the indicated arrow using ImageJ. Scale bars are 50 μm. (E-F) CRLF3 TAP (E) and control (F) iPSC MKs were lysed and immunoprecipitated (IP) with antibodies against FLAG, MOB1, and immunoglobulin G (IgG). Precipitated lysates were then probed for STK38, MOB1, and FLAG by western blot. (G) In vitro cultured MKs were seeded onto fibrinogen-coated coverslips and incubated at 37°C for 5 hours to induce proplatelet formation. Samples were fixed, stained for MOB1 (green), α-tubulin (red), and DAPI (blue), and imaged by fluorescence microscopy. Images are representative for Crlf3−/− and control (WT) proplatelet-forming MKs. (H) Western blot of in vitro cultured MKs probed with antibodies against pMOB1, MOB1, and GAPDH (left panel; n = 8 MOB1/GAPDH and 4 pMOB1/GAPDH) and pSTK38, STK38 and GAPDH (right panel; n = 3 STK38/GAPDH, 3 Crlf3−/−, and 4 WT pSTK38/GAPDH). (I) Three-dimensional structure of CRLF3 (residue 174 to end) solved by experimental phasing. Domains are labeled. Molecular graphics prepared using PyMOL. Data represent means ± standard deviations. Unpaired 2-tailed Student t test. *P < .05. FITC, fluorescein isothiocyanate; FN3, fibronectin type 3.
Finally, we sought to determine the crystal structure of CRLF3. We were successful only in expressing the C-terminal portion of CRLF3 (amino acids 174-442) at sufficient levels for crystallography. Crystals were successfully obtained using the sitting drop vapor diffusion method. Native data were collected to a resolution of 1.61 Å, and the structure was solved by Hg single-wavelength anomalous diffraction phasing. This revealed a 3-dimensional structure containing 2 known protein-binding domains, a fibronectin type 3 domain (residues 179-273) and a SPRY domain (residues 274-442; Figure 4I). Crystallographic statistics can be found in supplemental Table 3. The native structure, refined to Rwork = 0.177 and Rfree = 0.201, has been deposited at the Worldwide Protein Data Bank with ID 6RPX.
CRLF3 in human thrombopoiesis
We used a genetic approach to look for evidence that CRLF3 and its partners play a role in human thrombopoiesis. Using the imputed genotype data from 403 112 European ancestry participants in UK Biobank, we performed univariable association analyses between 29 hematological traits and genetic variants in the loci containing CRLF3, MOB1A, and STK38. Our analyses identified significant (−log10P > 8.08) associations with platelet distribution width in the CRLF3 locus (Figure 5A, left panel), of which the variant with the strongest evidence for association (rs6505211; purple diamond) was in the gene body. This variant was in high linkage disequilibrium (r2 > 0.8) with the variant exhibiting the strongest evidence for association with platelet distribution width but also with lymphocyte percentage of total white blood cells. We also identified associations with variants in STK38, which were significantly associated with mean platelet volume (Figure 5A, right panel) and identified a variant in MOB1A significantly associated with platelet count (supplemental Table 4). The latter variant is not associated with other hematological traits. These data therefore suggest a role for all 3 genes in thrombopoiesis, without necessarily implying that there is a mechanistic link among them.
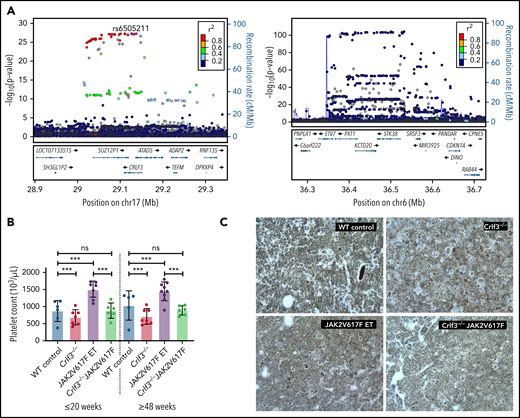
CRLF3 regulates platelet traits in humans and is a therapeutic target for ET. (A) Locuszoom of CRLF3 (left) and STK38 (right) showing variants associated with platelet distribution width (PDW) and mean platelet volume, respectively. The conditionally independent variant is indicated by a purple diamond; linkage disequilibrium (LD) values (r2) with this variant are indicated by dot colors according to the legend. The CRLF3 locuszoom plot shows the conditionally independent variant (rs6505211: −log10P, 27.1; MAF, 17.6%) is in high LD with a number of variants, which are significantly associated with PDW. In the case of STK38, the locuszoom plot indicates that the conditionally independent variant (rs141301223: −log10P, 10.4; MAF, 0.041%) is not in high LD with nearby variants (common for rare variant associations). (B) Platelet counts from young (≤20 weeks) and old (≥48 weeks) female WT control (blue; n = 6 young and 5 old), Crlf3−/− (red; n = 7 young and 7 old), JAK2 V617F ET (purple; n = 7 young and 9 old), and Crlf3−/− JAK2 V617F (green; n = 6 young and 6 old) mice. (C) Fixed tibia sections stained with Gömöri reticulin silver stain and imaged by light microscopy at 20× magnification. Images are representative of 3 mice per genotype. Data represent means ± standard deviations. Two-way analysis of variance with correction for multiple comparisons using the Holm-Sidak method. ***P < .005. ns, not significant.
CRLF3 regulates platelet traits in humans and is a therapeutic target for ET. (A) Locuszoom of CRLF3 (left) and STK38 (right) showing variants associated with platelet distribution width (PDW) and mean platelet volume, respectively. The conditionally independent variant is indicated by a purple diamond; linkage disequilibrium (LD) values (r2) with this variant are indicated by dot colors according to the legend. The CRLF3 locuszoom plot shows the conditionally independent variant (rs6505211: −log10P, 27.1; MAF, 17.6%) is in high LD with a number of variants, which are significantly associated with PDW. In the case of STK38, the locuszoom plot indicates that the conditionally independent variant (rs141301223: −log10P, 10.4; MAF, 0.041%) is not in high LD with nearby variants (common for rare variant associations). (B) Platelet counts from young (≤20 weeks) and old (≥48 weeks) female WT control (blue; n = 6 young and 5 old), Crlf3−/− (red; n = 7 young and 7 old), JAK2 V617F ET (purple; n = 7 young and 9 old), and Crlf3−/− JAK2 V617F (green; n = 6 young and 6 old) mice. (C) Fixed tibia sections stained with Gömöri reticulin silver stain and imaged by light microscopy at 20× magnification. Images are representative of 3 mice per genotype. Data represent means ± standard deviations. Two-way analysis of variance with correction for multiple comparisons using the Holm-Sidak method. ***P < .005. ns, not significant.
Among a collection of 59 464 individuals comprising probands affected with rare disorders for whom human platelet ontology terms were available and their first-degree relatives, we identified 27 who were heterozygous for severe impact variants in CRLF3. None had human platelet ontology terms suggesting a hematological phenotype (supplemental Table 5). No homozygous individuals for severe impact variants were identified in this cohort. Five individuals (including 2 siblings) were identified who were homozygous for missense variants, but again, none had a hematological phenotype. The severe variants were shown to be low frequency in gnomAD.34,43 The missense variants were inputted into the crystal structure described. Ala279Val and Asn410Asp have MAFs of 2.4 × 10−5 and 4.7 × 10−4 in gnomAD, respectively, but are minor changes on the surface of the protein. Leu389Pro has an MAF of 15% and together with the last variant, Thr392Ile, is part of a disordered loop (amino acids 387-398), again on the surface of the protein.
CRLF3 is a potential therapeutic target for ET
We postulated that the specific effect of CRLF3 deficiency on platelet count would make CRLF3 a potential therapeutic target in ET. As a proof of principle, we crossbred Crlf3−/− mice with a previously published inducible knock-in mouse model of ET driven by the JAK2 V617F mutation.44 The breeding strategy described in supplemental Figure 7 generated 4 groups of animals: WT control, Crlf3−/−, JAK2 V617F ET, and Crlf3−/− JAK2 V617F mice. We assessed the platelet counts in these 4 groups of mice at both young (≤20 weeks) and old (≥48 weeks) ages and showed that ablation of Crlf3 in JAK2 V627F ET mice normalized the platelet count to the levels seen in control mice (Figure 5B). Crucially, we showed that all other blood counts were unaffected (supplemental Table 6). Platelet function analysis in all 4 groups of mice showed no differences. No additional clinical findings were made in the Crlf3−/− JAK2V627F mice, including no evidence of BM fibrosis (Figure 5C) or changes in spleen size/weight (data not shown).
Discussion
Thrombopoiesis is classically described as a 2-stage process comprising first MK differentiation and maturation from HSCs followed by the actual process of platelet release. Proplatelet formation, which has been observed in vivo,1,2 is the broadly accepted mechanism by which MKs release platelets, although some authors have argued that MK fragmentation45 or membrane budding46 may constitute an alternative mechanism. Proplatelet fragments detach from MKs, forming long beads-on-a-string structures (preplatelets) that become mature discoid platelets.3 In this report, we show that preplatelet fission is critical for regulating platelet production and is the third and final stage of platelet production. We show that mice deficient in CRLF3 have reduced platelet counts resulting from ineffective thrombopoiesis, whereby slowed maturation of circulating preplatelets leads to their removal by the spleen, reducing the number of circulating platelets. These data clearly suggest the central role of proplatelet formation and subsequent preplatelet fission in platelet biogenesis as opposed to MK fragmentation or blebbing.
It has long been known that proplatelet formation is a process in which cytoskeletal proteins play a key role. Our data suggest that CRLF3 deficiency may exert its effect on preplatelet maturation through increasing tubulin stability. We show small changes in tubulin polyglutamylation in the primary mouse MKs, which may in part explain these observations. This would need to be confirmed in MKs derived from cell lines, rather than primary MKs, to allow for detailed protein and PTM studies to be carried out. We should note that it has been shown using cell line models that tubulin polyglutamylation promotes proplatelet-like extensions in CHO cells47 and affects the localization of motor protein in MKs.48 In keeping with this, we observed an increased rate of proplatelet formation in Crlf3−/− MKs. The tubulin C-terminal tail is subjected to diverse PTMs, which vary among cell type and intracellular localization.49 This allows fine spatial and temporal control of microtubule function by modifying the binding of microtubule-associated proteins (including microtubule-severing enzymes). Although the key enzymes involved in controlling tubulin glutamylation and severing are expressed equally between Crlf3−/− and control MKs (supplemental Table 7), we postulate that the increase in tubulin glutamylation observed in the Crlf3−/− MKs is such that it may subsequently affect tubulin severing in the Crlf3−/− preplatelets, thereby preventing their maturation. This hypothesis deserves further study, potentially using genetically modified MKs derived from cell lines enabling fine tuning of tubulin polyglutamylation to examine proplatelet formation and maturation dynamics.
Direct interaction between CRLF3 and MOB1 has been previously reported.41,42 MOB1 acts as a coactivator of NDR kinases, including STK38.38 MOB1 phosphorylation increases its binding to and activation of STK3839 and localizes the complex to the plasma membrane, especially at sites of pseudopodia/cytoplasmic extensions.50 MOB1 is known to affect tubulin stability through regulating acetylation and thereby cytokinesis.40 In this study, we confirmed that, in MKs, STK38 is associated with both MOB1 and CRLF3, but we did not show a direct interaction between CRLF3 and MOB1. However, MOB1 phosphorylation was increased in Crlf3−/− MKs, and we also showed that CRLF3 relocates to the membrane, but only in proplatelet-forming MKs. Taken together, these data strongly support the need for additional studies in MKs to provide definitive proof that MOB1 influences PTMs of tubulin in MKs and thereby influences proplatelet formation and maturation dynamics and ultimately circulating platelet numbers.
Most patients with ET are administered nonspecific cytoreductive therapies to lower their platelet count. Hydroxyurea, the most commonly prescribed agent, causes anemia, leukopenia, or skin ulcers51,52 in up to 20% of patients, and concerns about an associated leukemic risk remain.53,54 Anagrelide, the second most commonly prescribed agent, is associated with a threefold increased risk of myelofibrosis compared with hydroxyurea.55 Identification of a novel biological pathway that, when targeted, could specifically reduce platelet count is promising for the treatment of ET. Targeting CRLF3 may allow specific reductions in platelet count by acting at the level of preplatelet maturation, as evidenced with our Crlf3−/− JAK2 V617F ET murine model. These mice showed sustained and isolated normalization of their platelet count without increased BM fibrosis or leukemic transformation. An increased understanding of the role of CRLF3 in other cell types, its structure, and its structural relationship with partner proteins, such as STK38 and MOB1, as well as a definition of how CRLF3 interacts with tubulin after translation modifications, could potentially generate drugs that would complement (or supersede) the current nonspecific cytoreductive agents. Additional studies of the role of CRLF3 should also focus on the human system to confirm the findings of the murine studies presented here.
Acknowledgments
The authors acknowledge Tina Hamilton and Dean Pask for their expertise in and time with animal ethics and assistance with animal experiments. The authors also thank Jeremy Skepper for processing samples for scanning electron microscopy.
This work was supported by the British Heart Foundation (PhD Studentship FS/14/40/30921). This research was funded in part by the Wellcome Trust (203151/Z/16/Z) and the UKRI Medical Research Council (MC_PC_17230). For the purpose of open access, the authors have applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission. M.B. is supported by an Emmy Noether grant of the Deutsche Forschungsgemeinschaft (BE5084/3-1).
Authorship
Contribution: C.B. conceptualized the idea, designed and performed experiments, analyzed and interpreted data, and wrote manuscript; J.A.G. and M.L. conceptualized the idea, designed and performed experiments, analyzed and interpreted data, and revised the manuscript; S.S., A.K.W., Y.Y., R.W.M., J.B.-B., A.B., A.M., J.W., C.J.P., and P.A. designed and performed experiments and analyzed and interpreted data; L.M., T.M., A.L.E., S.M., G.J.H., and K.S.-P. designed experiments and interpreted data; D.J.A. conceptualized the idea and designed experiments; A.L.C. interpreted data and revised the manuscript; M.B. designed and performed experiments, analyzed and interpreted data, and revised the manuscript; W.N.E. performed experiments, interpreted data, and revised the manuscript; B.N. interpreted data and revised the manuscript; R.J.R. designed experiments, interpreted data, and revised the manuscript; and C.G. conceptualized the idea, designed experiments, interpreted data, and wrote the manuscript.
Conflict-of-interest disclosure: P.A. is a full-time employee of Regeneron pharmaceuticals and currently holds options and stock of the company. The remaining authors declare no competing financial interests.
Correspondence: Cavan Bennett, Cambridge Blood Centre, Long Road, Cambridge, CB20PT United Kingdom; e-mail: cavanbennett89@gmail.com; and Cedric Ghevaert, Wellcome-MRC Cambridge Stem Cell Institute, Jeffrey Cheah Biomedical Centre, Cambridge Biomedical Campus, University of Cambridge, Puddicombe Way, Cambridge CB2 0AW, United Kingdom; e-mail: cg348@cam.ac.uk.
CRLF3 structures have been deposited in the Worldwide Protein Data Bank under accession numbers 6RPX, 6RPY, and 6RPZ. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD017026. For original data, please contact cg384@cam.ac.uk.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal