

Key Points
GPR34 mutations in SG-MALT-lymphoma enhance the receptor signaling, particularly in the presence of ligand stimulation.
LELs enable the production of GPR34 ligand, hence providing paracrine stimulation to malignant B cells via GPR34.

Abstract
GPR34 translocation and mutation are specifically associated with salivary gland MALT lymphoma (SG-MALT-lymphoma). The majority of GPR34 mutations are clustered in its C-terminus, resulting in truncated proteins lacking the phosphorylation motif important for receptor desensitization. It is unclear why GPR34 genetic changes associate with SG-MALT-lymphoma and how these mutations contribute to the development of lymphoma. We generated isogenic Flp-InTRex293 cell lines that stably expressed a single copy of GPR34 or its various mutants and performed a range of in vitro assays. We found that the GPR34 Q340X truncation, but not the R84H and D151A mutants, conferred a significantly increased resistance to apoptosis and greater transforming potential than the GPR34 wild type. The GPR34 truncation mutant had a significantly delayed internalization compared with the wild type after ligand (lysophosphatidylserine) stimulation. Among the 9 signaling pathways examined, the GPR34 Q340X truncation, and to a lesser extent the D151A mutant, significantly activated CRE, NF-κB, and AP1 reporter activities, particularly in the presence of ligand stimulation. We further described the enhanced activities of phospholipase-A1/2 in the culture supernatant of Flp-InTRex293 cells that expressed the GPR34 Q340X mutant, as well as their potential to catalyze the synthesis of lysophosphatidylserine from phosphatidylserine. Importantly, phospholipase-A1 was abundantly expressed in the duct epithelium of salivary glands and those involved in lymphoepithelial lesions (LELs). Our findings advocate a model of paracrine stimulation of malignant B cells via GPR34, in which phospholipase A is released by LELs and hydrolyzes the phosphatidylserine exposed on apoptotic cells, generating lysophosphatidylserine, the ligand for GPR34. Thus, GPR34 activation potentially bridges LELs to genesis of SG-MALT-lymphoma.
Introduction
Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma) is a group of low grade B-cell lymphomas that share similar clinical, histologic, and immunophenotypic features and are thus considered as a single entity. The lymphoma originates from the marginal zone B cells of the acquired MALT, which are triggered by distinct chronic inflammatory disorders at different mucosal sites. Accordingly, MALT lymphomas of different sites exhibit remarkable divergences in their etiology and genetic makeup.1 For example, gastric MALT lymphoma commonly arises from a background of chronic Helicobacter pylori infection. The tumor growth in the majority of cases critically depends on H pylori–mediated immune responses, particularly T-cell help, whereas in the remaining cases, the minority is driven by a MALT1- or BCL10-involved chromosome translocation.2-4 In contrast, salivary gland MALT lymphoma commonly derives from a background of Sjögren’s syndrome, and most of the lymphoma-derived immunoglobulins are autoreactive, often possessing rheumatoid activities,5,6 indicating a role of chronic B-cell receptor signaling in its development. Genetically, salivary gland MALT lymphoma exhibits a mutation profile distinct from other MALT lymphomas, featured by recurrent GPR34 translocation or mutations.7-10
GPR34 is involved by t(X;14)(p11;q32), which juxtaposes GPR34 to the IG heavy chain enhancer region, thus causing its overexpression.7-9 Thus far, the translocation has been found only in salivary gland MALT lymphoma, occurring in ∼3% of cases. Similarly, GPR34 mutations are almost exclusively seen in salivary gland MALT lymphoma but are more frequent, occurring in 16% of cases.10 The majority of these mutations are nonsense changes or frameshift insertions and deletions that are clustered in the C-terminal region, resulting in truncated products lacking the C-terminal phosphorylation motif. The remaining mutations are missense changes, including R84H and D151A, with R84H and D151A affecting the tribasic (RKR) and highly conserved E/DRY motif, respectively. It is unclear why these genetic changes are specifically associated with salivary gland MALT lymphoma and how these genetic changes contribute to the lymphoma development at a molecular level.
GPR34 is a member of the class A G protein–coupled receptor (GPCR) superfamily, and its ligand has been identified as lysophosphatidylserine (LysoPS).11,12 LysoPS is generated by phospholipase A (PLA)-catalyzed hydrolysis of phosphatidylserine (PS), which is rich in the inner leaflet of the cell membrane but exposed during apoptosis.11,13 Ligand stimulation activates GPCR, triggering intracellular signaling through G proteins. By interacting with different G proteins, GPCR can activate diverse signaling pathways critical for cellular function.12,14 The receptor signaling is tightly regulated through a process known as desensitization that is triggered by phosphorylation at its C-terminal cytoplasmic tail, allowing its binding to β-arrestin and internalization and consequently dampening its receptor signaling.15
To understand the oncogenic action of various GPR34 mutations, we investigated their effects on the receptor internalization, intracellular signaling, and transformation potential in vitro. We also investigated the expression of PLA and identified evidence of paracrine stimulations via GPR34, independent of its mutation status, in salivary gland MALT lymphoma.
Materials and methods
GPR34 expression constructs
The full-length coding sequence of GPR34 in pLex-MCS (a gift from Anne Novak, Mayo Clinic) was used to generate various GPR34 mutants (R84H, D151A, and Q340X) by polymerase chain reaction and site-direct mutagenesis (Figure 1A). The wild-type and mutant GPR34 sequences were then cloned into both pcDNA5/FRT (supplemental Figure 1) and pIRESpuro vectors separately by using an NEBuilder HiFi DNA Assembly Cloning Kit (New England BioLabs); their sequences were confirmed by using Sanger sequencing. All the aforementioned constructs had a hemagglutinin (HA)-tag at the N-terminus of GPR34, and a separate set of these constructs with an additional C-terminal GFP-tag was also generated for analysis of GPR34 cellular localization.
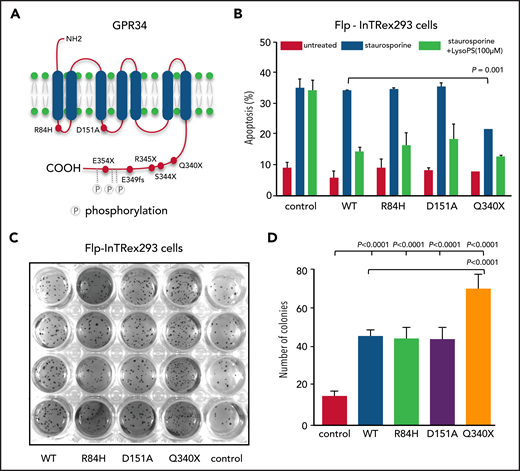
GPR34 truncation mutant confers resistance to apoptosis and transforming capacity. (A) Schematic illustration of GPR34 mutations seen in salivary gland MALT lymphoma. (B) Effect of GPR34 mutations on apoptosis induced by staurosporine. Isogenic Flp-InTRex293 cells that stably express a single copy of GPR34 or its various mutants were treated with 1.5 nM staurosporine in the absence or presence of LysoPS stimulation for 20 hours, and apoptosis was then measured by flow cytometry analysis of annexin V binding. Data are shown as mean ± standard deviation from 3 independent experiments. Statistical significance was analyzed by a two‐tailed unpaired Student t test, with significance indicated. (C) Transforming potential of GPR34 mutants determined by a soft agar colony formation assay. Isogenic Flp-INTRex293 cells that express a single copy of GPR34 or its various mutants were grown on soft agar for 3 weeks, and colonies were stained with a crystal violet and quantified. (D) Data are shown as mean ± standard deviation from 3 independent experiments. Statistical significance was analyzed by one‐way analysis of variance, with significance indicated. Control AQ19: the parental Flp-In T-REx-293 cell line. Wild-type (WT) or various mutations indicated: the derived stable expression cell line with single copy of WT or mutant GPR34.
GPR34 truncation mutant confers resistance to apoptosis and transforming capacity. (A) Schematic illustration of GPR34 mutations seen in salivary gland MALT lymphoma. (B) Effect of GPR34 mutations on apoptosis induced by staurosporine. Isogenic Flp-InTRex293 cells that stably express a single copy of GPR34 or its various mutants were treated with 1.5 nM staurosporine in the absence or presence of LysoPS stimulation for 20 hours, and apoptosis was then measured by flow cytometry analysis of annexin V binding. Data are shown as mean ± standard deviation from 3 independent experiments. Statistical significance was analyzed by a two‐tailed unpaired Student t test, with significance indicated. (C) Transforming potential of GPR34 mutants determined by a soft agar colony formation assay. Isogenic Flp-INTRex293 cells that express a single copy of GPR34 or its various mutants were grown on soft agar for 3 weeks, and colonies were stained with a crystal violet and quantified. (D) Data are shown as mean ± standard deviation from 3 independent experiments. Statistical significance was analyzed by one‐way analysis of variance, with significance indicated. Control AQ19: the parental Flp-In T-REx-293 cell line. Wild-type (WT) or various mutations indicated: the derived stable expression cell line with single copy of WT or mutant GPR34.
Generation of a single-copy GPR34 stable expression isogenic cell lines
Flp-InTRex293 host cells (a gift from Yvonne Vallis, Medical Research Council Laboratory of Molecular Biology, Cambridge) were cotransfected with each of the aforementioned GPR34 pcDNA5/FRT expression vectors together with the recombinase-producing pOG44 plasmid at a 1:9 ratio using TransIT-LT1 transfection reagent (Mirus) (supplemental Figure 1, available on the Blood Web site). The cells were cultured in Opti-MEM Reduced-Serum medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and 3 μg/mL blasticidin for 24 hours, and then subjected to hygromycin selection (150 μg/mL) in Dulbecco’s modified Eagle medium (DMEM) for 3 weeks. The resulting cell colonies were screened by polymerase chain reaction of the recombination site, and positive colonies were expanded and maintained in DMEM containing 100 μg/mL hygromycin and 3 μg/mL blasticidin. GPR34 expression in positive colonies (at least 3 for each construct) was further confirmed by western blot and/or flow cytometry analysis using an anti-HA antibody (supplemental Figure 2).
Impact of GPR34 expression on apoptosis
The Flp-InTRex293 cells (1 × 106) that carried a single copy of the wild-type or various mutant GPR34 expression constructs were treated with 1.5 nM staurosporine in the presence of LysoPS (100 μM) (carried by 4 mg/mL fatty acid free bovine serum albumin) or vehicle for 20 hours, grown on a glass surface in a 24-well culture dish. Apoptosis activities were measured by flow cytometry analysis of annexin V binding (eBioscience) and analyzed by using FlowJo 10 software.
Soft agar colony formation assay
The Flp-InTRex293 cells (1250) that carried a single copy of the wild-type or various mutant GPR34 expression construct, together with parental cells, were grown on standard soft agar containing 0.3% agarose and 1 × DMEM in a 24-well culture dish. The cells were fed twice a week and cultured for 3 weeks. Cell colonies were visualized by staining with crystal violet and quantified.
Analysis of GPR34 internalization
Analysis of GPR34 internalization was performed by using both time-lapse microscopy and flow cytometry. For time-lapse microscopy, the Flp-InTRex293 cells that carried a single copy of wild-type or various mutant GPR34 constructs with C-terminal GFP were seeded in a glass bottom dish (MatTek Life Science) for 24 hours. The cells were then treated with LysoPS (100 µM) in FluoroBrite medium (Gibco) while video-recorded for 30 minutes. The GPR34 expression was quantified by using ImageJ software (https://theolb.readthedocs.io/en/latest/imaging/measuring-cell-fluorescence-using-imagej.html).
For flow cytometry analysis, the Flp-InTRex293 cells that carried a single copy of wild-type or various mutant GPR34 expression constructs with an N-terminal HA-tag were suspended in a fasting medium at a density of 3 × 106 cells/mL. The cells were treated with LysoPS (100 µm), and an aliquot (100 µL) was taken out at the indicated times and analyzed by flow cytometry. Further details are given in the supplemental Methods.
Dual luciferase reporter assay
The firefly reporter plasmids for CRE (cAMP/PKA), SRF-RE (RhoA), SRE (MAPK/ERK), NFAT-RE (calcium/calcineurin), TCF/LEF-RE (Wnt), and ISRE (JAK/STAT1/2) were generated by cloning the corresponding synthetic response element sequences (Thermo Fisher Scientific) into the KpnI/HindIII cloning sites (supplemental Table 1). AP1 (MAPK/JNK), NF-κB, and CSL (NOTCH) reporter plasmids were from our previous studies.16,17 Each of these reporter assays was systematically optimized before data collection. Details regarding methods are given in the supplemental Methods.
Prediction of coupling probabilities of GPR34
G protein α coupling probability of GPR34 and its various mutants was estimated by using an online machine-learning program, PRECOG (predicting coupling probabilities of G-protein coupled receptors) (http://precog.russelllab.org).18 Details are given in the supplemental Methods.
PLA activity measurement
PLA2 activity was measured by using the EnzChek Phospholipase A2 Assay Kit (Thermo Fisher Scientific) (supplemental Figure 4). Because there is no commercial assay specific for PLA1 activity with PS as a substrate, we investigated the combined PLA1 and PLA2 activities by measuring free fatty acid release from the substrate 16:0-18:1 PS (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine) (Abcam). Details are given in the supplemental Methods.
Western blot analysis and immunohistochemistry
Western blot analysis was performed for PLA1, and immunohistochemistry was performed for GPR34, PLA1, and caspase 3. Details are given in the supplemental Methods. Local ethical guidelines were followed for the use of archival tissue materials for research, with the approval of the ethics committees of the involved institutions (05-Q1604-10).
Results
GPR34 mutants confer resistance to apoptosis
To investigate whether mutant GPR34 conferred resistance to apoptosis, we treated the isogenic Flp-InTRex293 cell lines with staurosporine and measured apoptosis by flow cytometry analysis of annexin V binding. In the absence of LysoPS stimulation, the cells expressing the GPR34 truncation mutant exhibited a significantly lower level of apoptosis than those expressing the wild type (Figure 1B). There was no difference in the level of apoptosis between the cell lines expressing the GPR34 missense mutants (R84H or D151A) and those expressing the wild type. In the presence of LysoPS stimulation, all the isogenic Flp-InTRex293 cell lines exhibited a similar reduction of apoptosis irrespective of expression of the wild-type or mutant GPR34, and all were significantly lower than the parental control cells. This finding suggests a pathogenic role for active receptor stimulation in addition to genetic changes.
GPR34 mutants exhibit enhanced transforming potential
To test whether mutation potentiates the transforming ability of GPR34, we performed a soft agar colony formation assay using the isogenic Flp-InTRex293 cells expressing the wild-type or various mutant GPR34 expression constructs. Compared with the parental cell line, the cells expressing the wild type and each of the GPR34 mutants exhibited a significantly higher number of colony formation, particularly those expressing the GPR34 truncation mutant (Figure 1C-D).
GPR34 mutants exhibit delayed internalization
Receptor internalization is a cardinal mechanism that desensitizes GPCR signaling after ligand stimulation. To investigate the impact of mutation on GPR34 internalization, we first generated isogenic Flp-InTRex293 cell lines that carried a single copy of the wild-type or various mutant GPR34 expression constructs. These isogenic cells were treated with the GPR34 ligand LysoPS (100 µM), and GPR34 expression was monitored by using time-lapse microscopy. All cell lines showed a rapid GPR34 internalization and degradation after ligand stimulation irrespective of the GPR34 mutation status (Figure 2A; supplemental Figure 5). However, the GPR34 truncation mutant exhibited a low level of membrane retention postligand stimulation, significantly higher than the wild type. Intriguingly, there was no difference between the GPR34 missense mutants (R84H and D151A) and the wild type (Figure 2B).
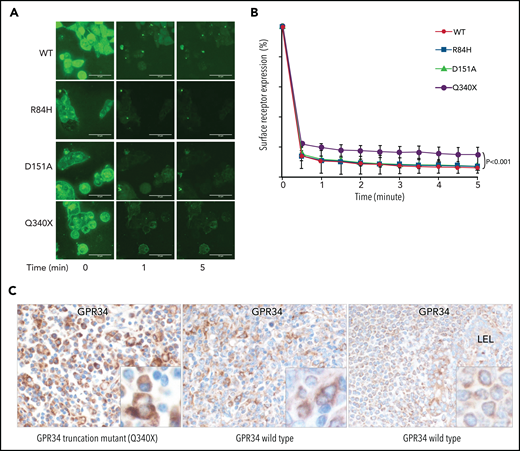
Ligand-induced GPR34 internalization is impaired by mutation. (A) Analysis of subcellular localization of GPR34 and its various mutants by using time-lapse microscopy. Isogenic Flp-InTRex293 cell lines that stably express a single copy of GPR34-GFP or its various mutants were treated with LysoPS (100 µM), and GPR34 expression was monitored over a 30-minute period. Shown is GPR34 expression at selected time points after ligand stimulation. The video record is presented in supplemental Figure 5. (B) The membrane expression in individual cells was quantified at the indicated time by using ImageJ software and normalized to the 0 time point. Comparison between wild-type (WT) GPR34 and its various mutants was performed with Prism 6 nonlinear regression analyses (GraphPad Software), with significant differences indicated. (C) GPR34 immunohistochemistry shows examples of strong (left), moderate (middle), and weak (right) staining.
Ligand-induced GPR34 internalization is impaired by mutation. (A) Analysis of subcellular localization of GPR34 and its various mutants by using time-lapse microscopy. Isogenic Flp-InTRex293 cell lines that stably express a single copy of GPR34-GFP or its various mutants were treated with LysoPS (100 µM), and GPR34 expression was monitored over a 30-minute period. Shown is GPR34 expression at selected time points after ligand stimulation. The video record is presented in supplemental Figure 5. (B) The membrane expression in individual cells was quantified at the indicated time by using ImageJ software and normalized to the 0 time point. Comparison between wild-type (WT) GPR34 and its various mutants was performed with Prism 6 nonlinear regression analyses (GraphPad Software), with significant differences indicated. (C) GPR34 immunohistochemistry shows examples of strong (left), moderate (middle), and weak (right) staining.
Similarly, flow cytometry analysis of these isogenic cell lines also revealed a significantly higher level of surface GPR34 for the truncation mutant than for the wild type after LysoPS stimulation (supplemental Figure 6). Again, there was no apparent difference in surface GPR34 expression between the missense mutants and the wild type.
GPR34 expression in salivary gland MALT lymphoma
To further investigate whether GPR34 mutation affects its protein expression, we performed GPR34 immunohistochemistry in 19 cases of salivary gland MALT lymphoma with known GPR34 mutation status from a previous study.10 Among the 5 cases of salivary gland MALT lymphomas that carried a nonsense (n = 3) or frameshift (n = 1) mutation in the C-terminus of GPR34 or a GPR34 translocation (n = 1), each exhibited strong GPR34 immunostaining in the lymphoma cells (Figure 2C). One case harbored the D151A change and showed weak GPR34 staining in the lymphoma cells. The remaining 13 cases were GPR34 mutation negative and exhibited variable staining intensities in lymphoma cells, ranging from strong (n = 4), moderate (n = 1), weak (n = 3), to negative (n = 5).
GPR34 mutants exhibit enhanced signaling capacity
GPCR can activate multiple cellular signaling pathways through its interacting G proteins. To investigate which signaling pathway is activated by GPR34 and how this is affected by mutation, we examined the activities of 9 effectors/signaling pathways downstream of G proteins using a dual luciferase reporter assay. In the absence of ligand stimulation, the wild-type GPR34 showed a low level of luciferase activity for the CRE (cAMP/PKA), NF-κB, AP1 (MAPK/JNK), SRF-RE (RhoA), and SRE (MAPK/ERK) reporters compared with control (Figure 3). Interestingly, the GPR34 Q340X truncation exhibited a significant increase in CRE (cAMP/PKA), NF-κB, and AP1 (MAPK/JNK) reporter activities; to a lesser extent, the D151A missense mutant exhibited an increase in the latter 2 reporter activities, compared with the wild-type GPR34, suggesting their constitutive activation. In the presence of LysoPS stimulation, the wild-type GPR34 showed a moderate increase in these reporter activities, whereas the GPR34 truncation (Q340X) displayed significantly enhanced reporter activities for CRE (cAMP/PKA), NF-κB, and AP1 (MAPK/JNK) and the D151A missense mutant for the latter 2 pathways, indicating their increased sensitivity to the ligand stimulation.
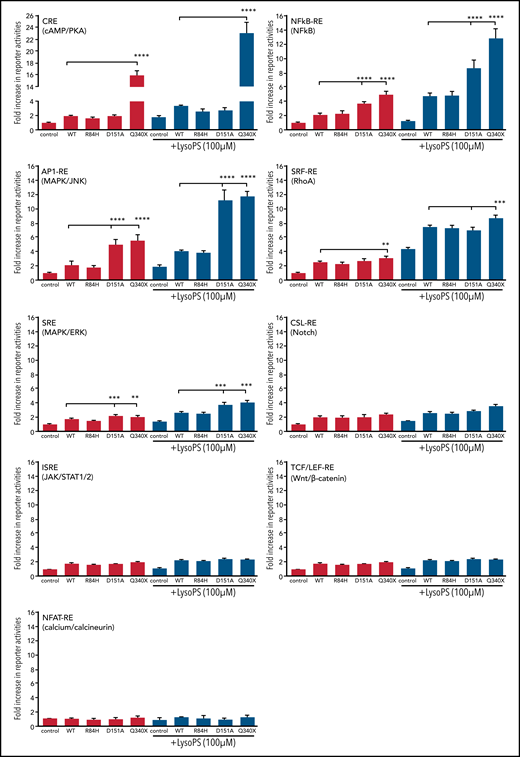
Comparison of GPR34 and its various mutants in activation of various signaling pathways using reporter assays. This was performed in isogenic Flp-InTRex293 cell lines that stably express a single copy of GPR34 or its mutant. Data (mean ± standard deviation) presented are from 3 independent experiments. In each reporter assay, the data are normalized to the reference control without LysoPS stimulation. Comparisons between various groups were assessed with one‐way analysis of variance with significant differences indicated (*P < .05; **P < .01; ***P < .001; ****P < .0001). Control: the parental Flp-In T-REx-293 cell line. Wild-type (WT) or various mutations indicated: the derived stable expression cell line with single copy of WT or mutant GPR34.
Comparison of GPR34 and its various mutants in activation of various signaling pathways using reporter assays. This was performed in isogenic Flp-InTRex293 cell lines that stably express a single copy of GPR34 or its mutant. Data (mean ± standard deviation) presented are from 3 independent experiments. In each reporter assay, the data are normalized to the reference control without LysoPS stimulation. Comparisons between various groups were assessed with one‐way analysis of variance with significant differences indicated (*P < .05; **P < .01; ***P < .001; ****P < .0001). Control: the parental Flp-In T-REx-293 cell line. Wild-type (WT) or various mutations indicated: the derived stable expression cell line with single copy of WT or mutant GPR34.
In contrast, the GPR34 R84H mutant exhibited no apparent difference from the wild type in these reporter activities regardless of LysoPS stimulation. GPR34 had little effect on the ISRE, TCF/LEF-RE, or NFAT-RE signaling pathway, irrespective of its mutation status (Figure 3).
Prediction of Gα coupling probabilities of GPR34
In general, there was a similar profile of Gα coupling probabilities among the wild-type GPR34 and its various mutants, as shown by the machine-learning program PRECOG. Nonetheless, the Q340X truncation mutant had a much higher binding probability to Gαi3, Gαo, and Gα15 than the wild type (Figure 4).
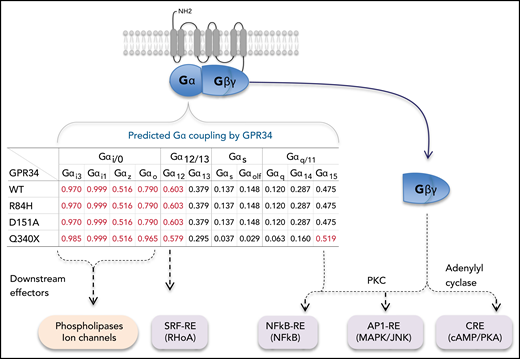
Prediction of Gα coupling probabilities of GPR34 and its various mutants by the machine-learning program PRECOG (http://precog.russelllab.org).18 A value >0.5 indicates a high probability of G protein–coupling property and is highlighted in red. The proposed effectors/signaling pathway activations downstream of Gα and Gβγ are based on previous literature.12,25–27 WT, wild type.
Prediction of Gα coupling probabilities of GPR34 and its various mutants by the machine-learning program PRECOG (http://precog.russelllab.org).18 A value >0.5 indicates a high probability of G protein–coupling property and is highlighted in red. The proposed effectors/signaling pathway activations downstream of Gα and Gβγ are based on previous literature.12,25–27 WT, wild type.
GPR34 mutants exhibit enhanced PLA activity
LysoPS is generated by hydrolysis of PS, which is catalyzed by PLA. PLA1 catalyzes the synthesis of 2-acyl-LysoPS, and PLA2 mediates the production of 1-acyl-LysoPS. Both 2-acyl-LysoPS and 1-acyl-LysoPS can activate GPR34, with the former being more potent.11 Because PLA is an effector molecule downstream of G-protein αi signaling,12 we investigated whether GPR34 activated PLA, which would in turn enhance LysoPS production.
Using a commercial assay, we first tested PLA2 activities in the isogenic Flp-InTRex293 cell line that carried a single copy of the wild-type or various mutant GPR34. Compared with the control, the cells expressing wild type and each of the mutant GPR34 exhibited significantly higher PLA2 activities, with the truncation mutant displaying the highest level (Figure 5A). The level of PLA2 activities also increased linearly with the substrate incubation time, suggesting potential autocrine stimulation.
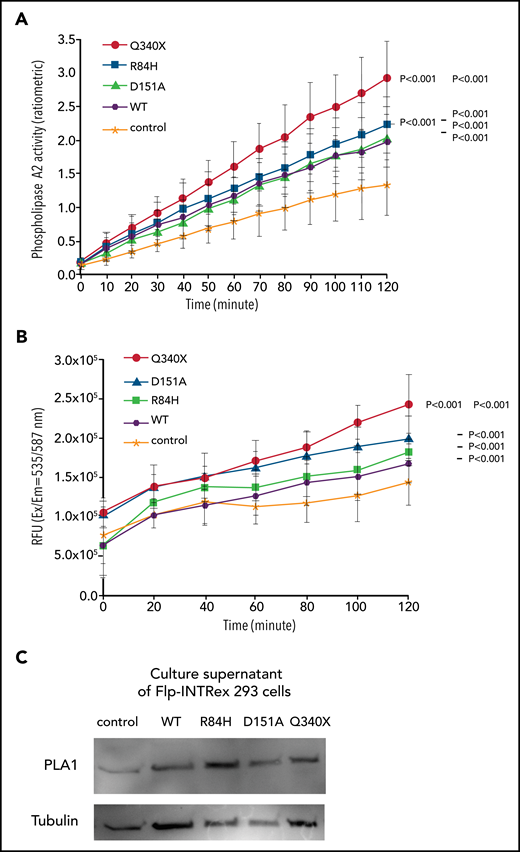
Measurement of PLA activities in Flp-INTRex293 cell culture. (A) PLA2 activity. Isogenic Flp-INTRex293 cells that express a single copy of GPR34 or its mutants, together with the parental cell line, were incubated with the substrate and liposome mix, and monitored for PLA2 enzymatic activity by using the EnzChek Phospholipase A2 Assay Kit. (B) Combined PLA1 and PLA2 activities. Isogenic Flp-INTRex293 cells that express a single copy of GPR34 or its mutants, together with the parental cell line, were incubated with 16:0-18:1 PS, and their culture supernatants were monitored for free fatty acid release at the indicated times by measuring their conversion to coenzyme A derivatives. Details are provided in the Materials and methods section for these assays. For each assay, the data (mean ± standard deviation) presented are from 3 independent experiments. Statistical differences among the various cell lines were analyzed by using a linear regression model, with significant differences indicated. (C) Confirmation of PLA1 in culture supernatant of Flp-INTRex293 cells by western blot analysis. Control: the parental Flp-In T-REx-293 cell line. Wild-type (WT) or various mutations indicated: the derived stable expression cell line with single copy of WT or mutant GPR34.
Measurement of PLA activities in Flp-INTRex293 cell culture. (A) PLA2 activity. Isogenic Flp-INTRex293 cells that express a single copy of GPR34 or its mutants, together with the parental cell line, were incubated with the substrate and liposome mix, and monitored for PLA2 enzymatic activity by using the EnzChek Phospholipase A2 Assay Kit. (B) Combined PLA1 and PLA2 activities. Isogenic Flp-INTRex293 cells that express a single copy of GPR34 or its mutants, together with the parental cell line, were incubated with 16:0-18:1 PS, and their culture supernatants were monitored for free fatty acid release at the indicated times by measuring their conversion to coenzyme A derivatives. Details are provided in the Materials and methods section for these assays. For each assay, the data (mean ± standard deviation) presented are from 3 independent experiments. Statistical differences among the various cell lines were analyzed by using a linear regression model, with significant differences indicated. (C) Confirmation of PLA1 in culture supernatant of Flp-INTRex293 cells by western blot analysis. Control: the parental Flp-In T-REx-293 cell line. Wild-type (WT) or various mutations indicated: the derived stable expression cell line with single copy of WT or mutant GPR34.
Because there is no commercial assay specifically for PLA1 activity with PS as substrate, we further investigated combined PLA1 and PLA2 activities by measuring the free fatty acids released from the substrate 16:0-18:1 PS after incubation with the isogenic Flp-InTRex293 cell lines. Compared with control, the cells expressing wild type and each of the GPR34 mutants exhibited significantly higher PLA activities, with the Q340X truncation mutant showing the highest level (Figure 5B).
These observations indicate that PLA was most likely released from the cells, thus capable of acting on the substrate provided. To investigate this theory, we performed western blot analysis of PLA1, as this is the major enzyme catalyzing the synthesis of LysoPS, the ligand for GPR34. PLA1 was found in the supernatant of each of the isogenic Flp-InTRex293 cell cultures (Figure 5C).
Evidence of autocrine stimulation of GPR34 in vitro
To further answer the speculation regarding autocrine stimulation, we tested whether incubation of GPR34 expressing Flp-InTRex293 cells with 16:0-18:1 PS activates GPR34 signaling using the luciferase reporter assay. The parental cells showed no apparent AP1-RE (MAPK/JNK) activity when incubated with PS. In contrast, Flp-InTRex293 cells expressing GPR34 Q340X mutant, and the wild type, displayed significantly increased AP1-RE reporter activity after incubation with PS (Figure 6). Because a phospholipid molecule with 2 acyl chains is unlikely capable of entering the ligand-binding pocket of the P2Y receptor as indicated by structural studies,19 the aforementioned observed AP1-RE reporter activities under PS incubation were most likely the result of stimulation by newly produced LysoPS.
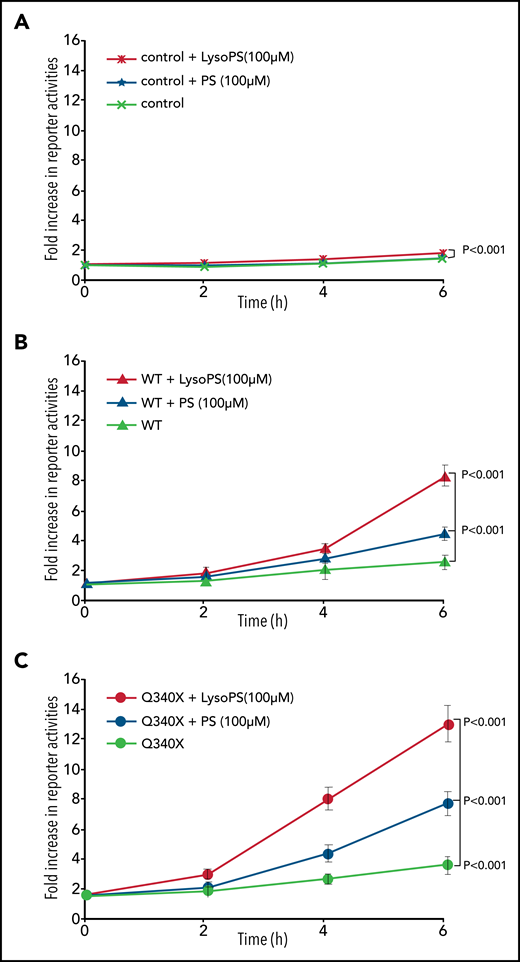
Autocrine activation of GPR34. Isogenic Flp-InTRex293 cell lines that stably express a single copy of GPR34 or its truncated variant, together with the parental cell lines (control), were transfected with AP1 firefly and Renilla reporter plasmids, cultured overnight, and then incubated with 16:0-18:1 PS or LysoPS for indicated times before measuring reporter activities. The data (mean ± standard deviation) presented are from 3 independent experiments. Statistical differences among different cell lines were analyzed by using a linear regression model, with significant differences indicated. WT, wild type.
Autocrine activation of GPR34. Isogenic Flp-InTRex293 cell lines that stably express a single copy of GPR34 or its truncated variant, together with the parental cell lines (control), were transfected with AP1 firefly and Renilla reporter plasmids, cultured overnight, and then incubated with 16:0-18:1 PS or LysoPS for indicated times before measuring reporter activities. The data (mean ± standard deviation) presented are from 3 independent experiments. Statistical differences among different cell lines were analyzed by using a linear regression model, with significant differences indicated. WT, wild type.
Potential paracrine stimulation of GPR34 in vivo
PLA1 is known to be expressed by digestive glands, and western blot analysis confirmed its expression in the normal pancreatic tissue and salivary gland biopsy specimens exhibiting myoepithelial sialadenitis (Figure 7A).
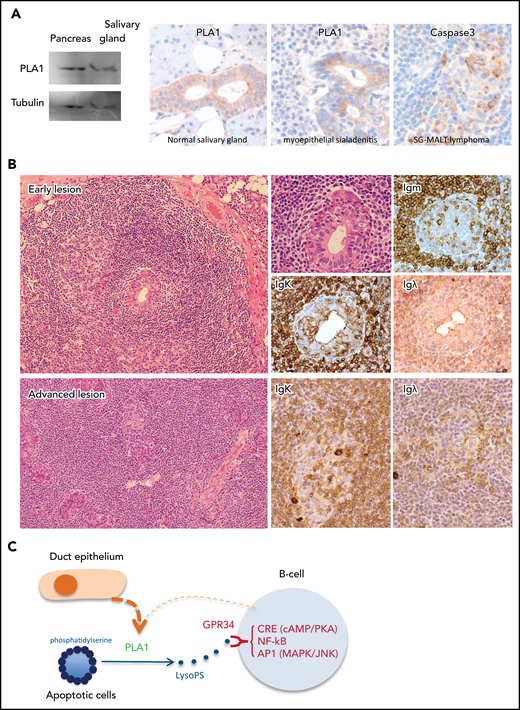
GPR34 activation by paracrine stimulation potentially bridges LELs to the genesis of salivary gland MALT lymphoma. (A) Left: western blot analysis shows PLA1 expression in a salivary gland biopsy specimen, with pancreatic tissue as a positive control. Middle: Immunohistochemistry shows PLA1 expression in the ductal but not acinar epithelium of adjacent normal salivary glands and those involved in LELs in a case of salivary gland MALT lymphoma. Right: Immunocytochemistry for cleaved caspase 3 shows prominent apoptotic activities in LELs in a salivary gland MALT lymphoma. (B) Histology of salivary gland MALT lymphoma. An early lesion shows emergence of neoplastic B cells surrounding the LELs, with some of the neoplastic B cells invading the epithelial gland, as indicated by the immunoglobulin K (IgΚ) light chain restriction. An advanced lesion displays diffuse lymphoid infiltration and prominent LELs with malignant B cells expanding around the LELs. (Images are courtesy of Peter G. Isaacson.) (C) GPR34 activation by paracrine stimulation: PLA1 released by duct epithelial cells of LELs can hydrolyze PS exposed on apoptotic cells, generating LysoPS and hence stimulating malignant B cells via GPR34.
GPR34 activation by paracrine stimulation potentially bridges LELs to the genesis of salivary gland MALT lymphoma. (A) Left: western blot analysis shows PLA1 expression in a salivary gland biopsy specimen, with pancreatic tissue as a positive control. Middle: Immunohistochemistry shows PLA1 expression in the ductal but not acinar epithelium of adjacent normal salivary glands and those involved in LELs in a case of salivary gland MALT lymphoma. Right: Immunocytochemistry for cleaved caspase 3 shows prominent apoptotic activities in LELs in a salivary gland MALT lymphoma. (B) Histology of salivary gland MALT lymphoma. An early lesion shows emergence of neoplastic B cells surrounding the LELs, with some of the neoplastic B cells invading the epithelial gland, as indicated by the immunoglobulin K (IgΚ) light chain restriction. An advanced lesion displays diffuse lymphoid infiltration and prominent LELs with malignant B cells expanding around the LELs. (Images are courtesy of Peter G. Isaacson.) (C) GPR34 activation by paracrine stimulation: PLA1 released by duct epithelial cells of LELs can hydrolyze PS exposed on apoptotic cells, generating LysoPS and hence stimulating malignant B cells via GPR34.
We further investigated PLA1 expression in normal salivary glands adjacent to salivary gland adenocarcinoma that showed no evidence of an underlying inflammatory condition according to histology (n = 7) or myoepithelial sialadenitis (n = 5) and salivary gland MALT lymphoma (n = 17 [8 with adjacent intact salivary glands]) by immunohistochemistry. Moderate PLA1 expression was seen in the ductal but not acinar epithelium of normal salivary glands adjacent to salivary gland adenocarcinoma or salivary gland MALT lymphoma (Figure 7A; supplemental Figure 7). In myoepithelial sialadenitis, PLA1 staining in lymphoepithelial lesions (LELs) appeared stronger than that in the normal ductal epithelium. Although PLA1 staining in LELs in salivary gland MALT lymphoma is variable, it is often weaker than that in the normal ductal epithelium. There was no detectable PLA1 expression in lymphoma B cells.
We also performed immunohistochemistry for cleaved caspase 3 in salivary gland MALT lymphoma (n = 17) and found active apoptosis in LELs in each of the specimens examined (Figure 7; supplemental Figure 7). Because apoptotic cells typically expose their inner membrane leaflet, which is rich in PS, the exposed PS could be hydrolyzed by PLA1 released from LELs, producing LysoPS, the ligand for GPR34.
Discussion
By functional characterization of the lymphoma-derived GPR34 mutations, we have shown in the current study that the C-terminal truncation and D151A mutations are activation changes, causing GPR34 constitutive activation and their hypersensitive responses to ligand stimulation. We also described expression of PLA1 in the duct epithelium of LELs in salivary gland MALT lymphoma, which could act on PS exposed on apoptotic cells and generate ligand for GPR34, providing paracrine stimulation to malignant B cells.
There are clear differences in the functional effects among the GPR34 mutations investigated, particularly between truncation and missense mutants. The GPR34 Q340X truncation mutant conferred resistance to apoptosis induced by staurosporine and higher transforming potential as measured by soft agar colony formation assay, whereas the 2 missense mutants exhibited no difference from the wild-type GPR34. The Q340X truncation mutant lacks the C-terminal phosphorylation motif,10 which is responsible for binding to β-arrestin, receptor desensitization, and internalization,15 and showed clear evidence of membrane retention after ligand stimulation, albeit at a low level. In contrast, the 2 missense mutants displayed no difference from the wild-type GPR34 in their receptor internalization and degradation. The difference in the internalization between the GPR34 truncation and missense mutants seen in the in vitro experiments is also supported by the observation of high expression of the truncation but not missense mutants in primary salivary gland MALT lymphoma cells. Nonetheless, the truncation mutant, as with the GPR34 wild type, also exhibited drastic receptor internalization immediately after the ligand stimulation, indicating the presence of an alternative mechanism of receptor internalization independent of phosphorylation motif/β-arrestin interaction.20
Among the GPR34 mutants investigated, the truncation mutant showed the highest capacity in activation of several intracellular signaling pathways. Importantly, the truncation mutant activated the CRE (cAMP/PKA), NF-κB, and AP1 signaling pathways in the absence of ligand stimulation, suggesting its constitutive activation. In addition, the truncation mutant was far more sensitive to ligand stimulation in activation of these signaling pathways than the missense mutants, in keeping with its impaired internalization and desensitization.
The GPR34 D151A mutant exhibited moderate capacity in activation of the NF-κB and AP1 signaling pathways, including in the absence of ligand stimulation, but displayed no impact on apoptosis and soft agar colony formation assays. These variant observations most likely reflect the differences in the sensitivity of these in vitro assays, with the reporter assay being more sensitive. GPR34 D151A affects the highly conserved E/DRY motif at the cytosolic end of the third transmembrane domain,10 which is critical for receptor activation and function. Replacement of the acidic amino acid (glutamic acid or aspartic acid) by a nonpolar amino acid such as alanine results in more efficient signaling properties in both the presence and absence of agonist stimulation.21-23 Taken together, GPR34 D151A is an activation change, although it appears less potent than the truncation mutation.
Surprisingly, the GPR34 R84H mutant showed no apparent difference from the wild type among the in vitro assays used in this study. This mutant affects the tribasic RKR motif in the first intracellular loop, which is the key topogenic signal determining the orientation of the first transmembrane domain.24 It is worth noting that the mutation is a conservative amino acid replacement, which may explain its lack of any major functional impact or why it was not detected by the functional assays used in this study.
Among the 9 intracellular signaling pathways investigated, the GPR34 truncation, and to a lesser extent the D151A mutant, mainly activates the CRE (cAMP/PKA), NF-κB, AP1, and SRF-RE signaling pathways. This signaling profile is identical to that of overexpressed GPR34 wild type established by using a similar reporter assay in a previous study,7 suggesting that mutation augments signaling activities without rerouting to different downstream signaling targets. In line with this, there is a similar G protein coupling profile between the GPR34 wild type and various mutants, as predicted by the online machine-learning program (PRECOG) (Figure 4). Interestingly, the Q340X truncation mutant had a much higher binding probability to Gαi3, Gαo, and Gα15 than the wild type, consistent with its gain-of-function properties as shown by the different in vitro assays. This predicted G protein coupling profile is broadly in line with the downstream signaling pathways activated by GPR34 as shown by reporter assays, with the exception of CRE (cAMP/PKA) or adenylyl cyclase. Among Gα family members, adenylyl cyclase is primarily activated by Gαs but inhibited by Gαi.12 However, there is now overwhelming evidence that the Gβγ units can also activate a variety of effectors, including adenylyl cyclase.25-27 Thus, the observed signaling activities by GPR34 mutants may be mediated by the Gβγ units.
One of the effector molecules downstream of G-protein αi signaling is PLA.12 We showed that the Flp-INTRex293 cells expressing GPR34 or its mutants, particularly the truncation mutant, had higher PLA activities than control cells. Although it remains to be confirmed whether PLA is expressed in malignant B cells, PLA1, the major enzyme for catalyzing LysoPS production, is abundantly expressed in the duct epithelium of salivary glands and also those involved in LELs. It is worth noting that the LELs in myoepithelial sialadenitis and salivary gland MALT lymphoma involve the duct, rather than the acini that lack PLA1 expression.
Salivary gland MALT lymphomas invariably arise from a background of myoepithelial sialadenitis and exhibit extensive LELs, with the lymphoma cells emerging and expanding around these lesions, giving rising to a halo appearance, which is an important feature for locating lymphoma cells (Figure 7B). The apoptotic cells in LELs may act as a source of PS as it is rich in the inner leaflet of the cell membrane and exposed during apoptosis. Hydrolysis of the exposed PS by PLA1 released or secreted by duct epithelial cells would produce LysoPS, the ligand for GPR34, thus providing paracrine stimulation to promote the survival and expansion of neoplastic B cells (Figure 7C). Importantly, LELs are a dynamic process, with relentless epithelial cell re-generation and apoptosis, thus providing a perpetual and renewal microenvironment for persistent stimulation of malignant B cells. Such paracrine stimulation may represent an important common mechanism in the pathogenesis of salivary gland MALT lymphoma independent of GPR34 genetic changes, although those with mutant or overexpressed GPR34 are more sensitive to the ligand stimulation. This elegantly explains why the emergence of clonal malignant B cells is first seen around LELs and also why the GPR34 mutation and translocation are preferentially associated with salivary gland MALT lymphoma, as the full oncogenic potential of these genetic changes still depends on active receptor stimulation.7-10 In keeping with this notion, both PLA1 and PLA2 activities are highly elevated in sera of patients with systemic lupus erythematosus and Sjögren’s syndrome, particularly in those with active disease.28,29
The B-cell receptor expressed by most if not all salivary gland MALT lymphomas is autoreactive, with the majority bearing rheumatoid factor–like activities.5,10 Chronic B-cell receptor signaling is also critical for the emergence and clonal expansion of transformed B cells and may thus cooperate with the GPR34 signaling in the development of salivary gland MALT lymphoma.
In summary, our findings indicate that GPR34 mutations are activating changes and advocate a model for paracrine stimulation of malignant B cells via GPR34 by ligand (LysoPS) independent of its mutation status, whose production is enabled by LELs. This mechanistic model clarifies the specific association between GPR34 translocation/mutation and salivary gland MALT lymphoma, and also provides a common mechanism explaining the emergence of neoplastic B cells and their expansion surrounding the LELs. The model also offers a molecular basis for the development of therapeutic strategies for both salivary gland MALT lymphoma and myoepithelial sialadenitis.
Acknowledgments
The authors thank Timothy Cox for help accessing their microplate reader and Ian Clark for help using their microscopy facility.
This research in M.-Q.D.’s laboratory was supported by grants from the Kay Kendall Leukemia Fund (KKL1141) UK, Blood Cancer UK (13006), Isaac Newton Trust, Addenbrooke’s Charitable Trust, and the Department of Pathology, University of Cambridge. The Human Research Tissue Bank is supported by the NIHR Cambridge Biomedical Research Centre.
Authorship
Contribution: B.K., D.K., W.Z., and M.-Q.D. designed experiments and collected and analyzed data; B.K. and M.-Q.D. prepared and wrote the manuscript; A.C.W. input pathology; M.-Q.D. designed and coordinated the study and contributed to research funding; and all authors commented on the manuscript and approved its submission for publication.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ming-Qing Du, Division of Cellular and Molecular Pathology, Department of Pathology, University of Cambridge, Box 231, Level 3, Laboratory Block, Addenbrooke’s Hospital, Hills Rd, Cambridge, CB2 2QQ, United Kingdom; e-mail: mqd20@cam.ac.uk.
Requests for original data may be submitted to the corresponding author (Ming-Qing Du; e-mail: mqd20@cam.ac.uk).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal