

Key Points
SOD2 V16A is associated with clinical markers of endothelial dysfunction in sickle cell patients.
SOD2 V16A drives increased reactive oxygen species through decreased mitochondria complex IV activity.

Abstract
Superoxide dismutase 2 (SOD2) catalyzes the dismutation of superoxide to hydrogen peroxide in mitochondria, limiting mitochondrial damage. The SOD2 amino acid valine-to-alanine substitution at position 16 (V16A) in the mitochondrial leader sequence is a common genetic variant among patients with sickle cell disease (SCD). However, little is known about the cardiovascular consequences of SOD2V16A in SCD patients or its impact on endothelial cell function. Here, we show SOD2V16A associates with increased tricuspid regurgitant velocity (TRV), systolic blood pressure, right ventricle area at systole, and declined 6-minute walk distance in 410 SCD patients. Plasma lactate dehydrogenase, a marker of oxidative stress and hemolysis, significantly associated with higher TRV. To define the impact of SOD2V16A in the endothelium, we introduced the SOD2V16A variant into endothelial cells. SOD2V16A increases hydrogen peroxide and mitochondrial reactive oxygen species (ROS) production compared with controls. Unexpectedly, the increased ROS was not due to SOD2V16A mislocalization but was associated with mitochondrial complex IV and a concomitant decrease in basal respiration and complex IV activity. In sum, SOD2V16A is a novel clinical biomarker of cardiovascular dysfunction in SCD patients through its ability to decrease mitochondrial complex IV activity and amplify ROS production in the endothelium.
Introduction
Although sickle cell disease (SCD) is ascribed to a single point mutation in β-globin gene, cardiovascular presentation among patients remains highly variable. For example, pulmonary hypertension occurs in 6% to 11% of adult SCD patients and predicts early death1,2 whereas 24% of patients exhibit signs of stroke by 45 years of age.3 Although several factors contribute to cardiovascular deviation among SCD patients, genetic variability and oxidative stress are key elements driving phenotype heterogeneity.
Mitochondria dysfunction contributes to SCD pathogenesis by increasing reactive oxygen species (ROS).4 Mitochondria uncoupling increases superoxide (O2•-) formation, which is managed by superoxide dismutase 2 (SOD2), a mitochondrial matrix protein that catalyzes dismutation of O2•- to hydrogen peroxide (H2O2). A valine-to-alanine substitution (SOD2V16A, rs4880) in the mitochondria leader sequence is found in 45% of people with African ancestry, 65% of Latinos, 54% of South Asians, and 52% of Europeans.5 In SCD, SOD2V16A associates with acute splenic sequestration and vaso-occlusive crises in children6 and increased risk of stroke in adults.7 However, the impact of SOD2V16A on cardiovascular severity remains unclear, and rigorous functional studies defining the impact of SOD2V16A remain elusive. This stems from contradictory results suggesting enzymatic activity of SOD2V16A can be increased8 or decreased.6 We hypothesized that SOD2V16A drives mitochondria dysfunction, increases mitochondrial ROS production, and can be used as a prognostic genetic biomarker for vasculopathy in SCD patients.
Study design
Cohort and genetic analysis
SCD patients from the Treatment of Pulmonary Hypertension and Sickle Cell Disease With Sildenafil Therapy (walk-PHaSST) trial were recruited for genetic analysis (approved under University of Pittsburgh Institutional Review Board Protocol STUDY19060255).9 Genotyping of rs4880 was performed using Taqman genotyping assay (C___8709053_10), and methods were performed as described and adjusted for age, gender, BMI, hemoglobin, pulse rate, antihypertensive treatment, α-globin deletion, and study site.10
HPAEC transduction and transfection with APEX
Polyhistidine-tagged human SOD2 rs4880 (A) encoding valine on the 16th amino acid position (SOD2WT) or SOD2 rs4880 (G) encoding alanine on the 16th amino acid position (SOD2V16A) was placed on a bicistronic lentiviral vector that also coexpressed green fluorescent protein (GFP) as previously described.11 Human pulmonary artery endothelial cells (HPAECs) were incubated with lentivirus and 1 ug/mL polybrene for 8 hours in a 6-well plate (250 000 cells per well) to achieve a twofold increase over endogenous SOD2. Experiments were performed 72 hours postlentiviral transduction. HPAECs were transfected with 0.5 µg SOD2WT or SOD2V16A conjugated with APEX2 using Nucleofector; experiments were conducted 24 hours posttransfection. APEX2 assays were performed as previously described.12
Statistics
Data were expressed as mean ± standard deviation from the mean unless otherwise specified. The n values represent biological replicates. A Student t test was used to determine significance using GraphPad Prism version 8 software. A value of P < .05 was considered significant.
Please see extended methods in the supplemental Material, available on the Blood Web site.
Results and discussion
To evaluate the impact of SOD2V16A on cardiovascular phenotypes, we genotyped 410 SS genotype SCD patients from the walk-PHaSST trial.9 Using multiple linear regression analysis across genotype, we found increased tricuspid regurgitant velocity (TRV, a marker of pulmonary hypertension), systolic blood pressure, and systolic right ventricular area (Figure 1A-C) along with decreased 6-minute walk distance (Figure 1D) correlated with the rs4880 genotype. These clinical parameters are suggestive of reduced pulmonary and cardiovascular function and heightened dysfunctional endothelium. A strong trend toward decreased hemoglobin concentration and a significant increase in the hemolytic index were observed, indicating a potential role for anemia and hemolysis (supplemental Table 1). Plasma lactate dehydrogenase LDH, a marker of hemolysis,13 also strongly correlated with TRV in rs4880G homozygous patients (Figure 1E). Extracellular activity of LDH increases due to cell membrane perturbation by lipid peroxidation.14 Thus, LDH is used as a clinical biomarker of oxidative stress and inflammation.13,14 Historically, elevated LDH levels in SCD patients associate with increased TRV and pulmonary hypertension.13 Although we did not observe increased LDH levels in rs4880G homozygotes (supplemental Table 1), these data suggest that rs4880G associates with worse cardiovascular outcomes in the presence of oxidative stress and inflammation. It is also possible that increased oxidative stress exacerbates hemolysis increasing TRV.
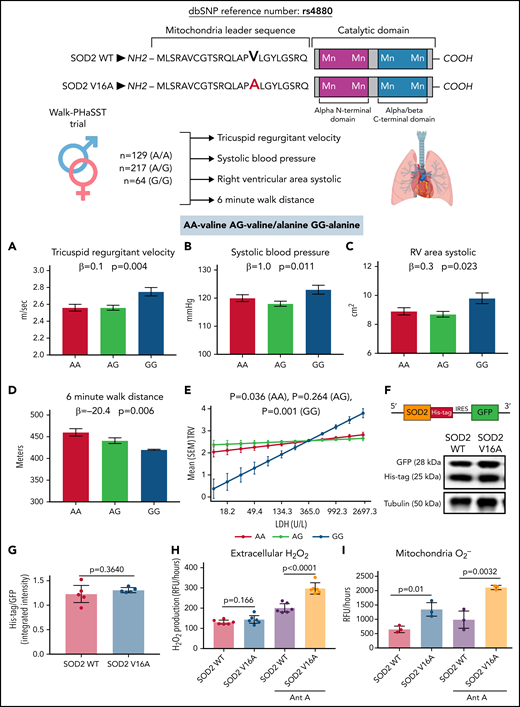
SOD2 V16A is associated with clinical markers of endothelial dysfunction and increases extracellular hydrogen peroxide and mitochondrial superoxide production. (A) Tricuspid regurgitant velocity (TRV), (B) systolic blood pressure, (C) right ventricular area at systole, (D) 6-minute walk distance of Walk-PhaSST cohort by SOD2 16th amino acid genotype, and (E) interaction between lactate dehydrogenase and TRV by SOD2 16th amino acid genotype P for interaction of LDH and genotype <.001. (F) Plasmid schematic of SOD2 lentiviral and (G) protein expression between 2 SOD2 variants. (H) Extracellular hydrogen peroxide and (I) mitochondrial reactive oxygen species produced by each SOD2 variant with and without antimycin A. Results in panels A-E are in mean with standard error of the mean. In panels A-D, β is a measure of change in outcome by each minor allele (additive model) and P values represent the test for trend. In panel E, interaction between SOD genotype and LDH was tested in a linear regression analysis. P values test the correlation between TRV and LDH in each SOD genotype. Results in panels G-I are given in mean ± standard deviation. AA represents patients with the alanine variant of SOD2, AG represents patients with the alanine/valine variant, and GG represents patients with the valine variant. LDH, lactate dehydrogenase.
SOD2 V16A is associated with clinical markers of endothelial dysfunction and increases extracellular hydrogen peroxide and mitochondrial superoxide production. (A) Tricuspid regurgitant velocity (TRV), (B) systolic blood pressure, (C) right ventricular area at systole, (D) 6-minute walk distance of Walk-PhaSST cohort by SOD2 16th amino acid genotype, and (E) interaction between lactate dehydrogenase and TRV by SOD2 16th amino acid genotype P for interaction of LDH and genotype <.001. (F) Plasmid schematic of SOD2 lentiviral and (G) protein expression between 2 SOD2 variants. (H) Extracellular hydrogen peroxide and (I) mitochondrial reactive oxygen species produced by each SOD2 variant with and without antimycin A. Results in panels A-E are in mean with standard error of the mean. In panels A-D, β is a measure of change in outcome by each minor allele (additive model) and P values represent the test for trend. In panel E, interaction between SOD genotype and LDH was tested in a linear regression analysis. P values test the correlation between TRV and LDH in each SOD genotype. Results in panels G-I are given in mean ± standard deviation. AA represents patients with the alanine variant of SOD2, AG represents patients with the alanine/valine variant, and GG represents patients with the valine variant. LDH, lactate dehydrogenase.
To investigate the impact of endothelial SOD2V16A, we coexpressed either valine or alanine SOD2 and GFP (Figure 1F) in HPAECs. Using GFP as an expression control, we found no difference in the exogenous SOD2 protein levels, suggesting that SOD2V16A did not affect protein stability (Figure 1F-G). Next, we tested whether SOD2V16A impacted enzymatic activity by measuring H2O2 production in HPAECs. Although there was no difference in H2O2 production at baseline, the SOD2V16A variant exacerbated antimycin A, a complex III inhibitor, induced H2O2 production compared with SOD2WT(Figure 1H). Inhibiting catalase failed to correct the differences in H2O2 levels between SOD2 variants (supplemental Figure 1A). Furthermore, SOD2V16A did not alter catalase or SOD1 protein expression (supplemental Figure 1B-C), suggesting that the difference in H2O2 production was independent of catalase and SOD1. These results indicate that SOD2V16A exacerbates H2O2 formation in HPAECs under stressed conditions.
Although enzymatic dismutation by SOD is the most efficient method of dismutation, O2•− can spontaneously dismutate to H2O2 independent of SOD.15 Therefore, we tested if increased mitochondrial O2•− production could be the source of increased H2O2. We found increased mitochondrial O2•− levels both at baseline and with antimycin A treatment in the SOD2V16A-transduced HPAECs (Figure 1I). Increased mitochondria O2•− could indicate compromised SOD2 activity. However, when inhibiting SOD1, the only other intracellular SOD isoform, we found SOD activity remained the same between the variants, indicating that SOD2V16A did not alter SOD2 activity (supplemental Figure 2A-B).
Because SOD2V16A retained normal activity, we hypothesized that SOD2V16A mislocalizes within HPAECs, considering that the V16A mutation resides in the mitochondria leader sequence. To examine SOD2 subcellular location, we fused APEX2, a peroxidase used for electron microscopy and identifying protein-protein interactions, to the C terminus of the 2 SOD2 variants.12 We observed no difference in localization between SOD2-APEX2 variants within the mitochondrial matrix (Figure 2A; supplemental Figure 2C). Because neither activity nor localization of SOD2V16A were affected, the most likely explanation for the elevated mitochondrial O2•− is enhanced production. The respiratory chain is the predominant source of mitochondrial O2•− generation. Therefore, we investigated whether SOD2V16A disrupted respiratory chain complex formation or activity. Although the protein expression of respiratory chain complexes was not affected by SOD2V16A (Figure 2B-C), the variant interacted significantly more with complex IV (Figure 2D-E). Specifically, SOD2V16A interacted more with subunit 2, part of the catalytic domain of complex IV, and showed significantly lower complex IV activity (Figure 2F). Inhibited complex IV activity impairs mitochondrial respiration.16 Indeed, we found that basal oxygen consumption was lower in SOD2V16A-expressing HPAECs, indicating suppressed mitochondrial respiration (Figure 2G). Additionally, there was a compensatory increase in glycolysis and fatty acid oxidation (supplemental Figure 2D-E).
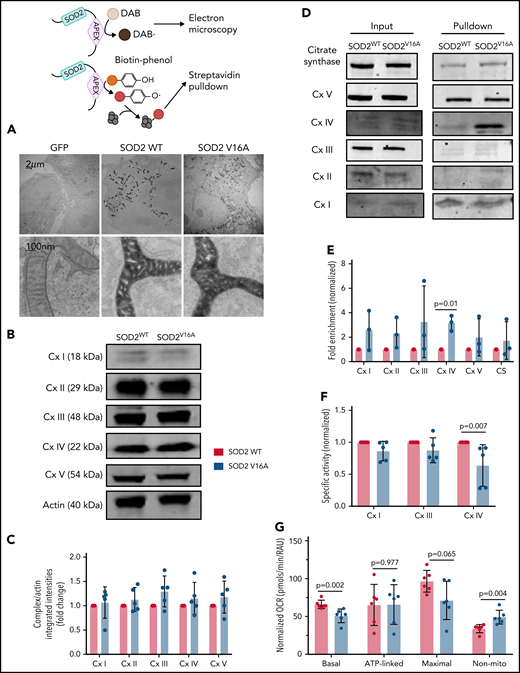
SOD2 V16A associates with complex IV of the mitochondria respiration chain, resulting in decreased complex IV activity and basal respiration in human arterial pulmonary endothelial cells. (A) Electron microscopy images of APEX-fused SOD2 variants obtained at ×100 000 magnification with JEM 1400Plus transmission electron microscope. (B-C) Western blots and quantification of mitochondria respiration chain complexes in SOD2 variants. (D-E) Western blots and fold enrichment calculations of APEX-fused SOD2 variants after biotinylating and streptavidin pulldown. (F) Enzymatic activities of complexes I, III, and IV in SOD2 variants normalized to complex V activity. (G) Mitochondria respiration of cells transduced with either SOD2 variant. Results are given in mean with standard deviation.
SOD2 V16A associates with complex IV of the mitochondria respiration chain, resulting in decreased complex IV activity and basal respiration in human arterial pulmonary endothelial cells. (A) Electron microscopy images of APEX-fused SOD2 variants obtained at ×100 000 magnification with JEM 1400Plus transmission electron microscope. (B-C) Western blots and quantification of mitochondria respiration chain complexes in SOD2 variants. (D-E) Western blots and fold enrichment calculations of APEX-fused SOD2 variants after biotinylating and streptavidin pulldown. (F) Enzymatic activities of complexes I, III, and IV in SOD2 variants normalized to complex V activity. (G) Mitochondria respiration of cells transduced with either SOD2 variant. Results are given in mean with standard deviation.
Despite the compromised complex IV activity, SOD2V16A-expressing HPAECs maintained normal adenosine triphosphate (ATP) production and mitochondrial potential (supplemental Figure 3A-B). However, we observed increased mitochondrial hyperfusion (supplemental Figure 3C-D). This is consistent with the previous observation that reduced complex IV activity results in mitochondria hyperfusion as a mechanism to maintain ATP production.17 These data suggest that SOD2V16A expressed in HPAECs preserve ATP production by hyperfusing mitochondria but does so at the expense of generating more mitochondrial O2•−. This increase in ROS production, however, did not result in a change in endothelial cell proliferation and migration, cell survival, or HIF-2α expression. However, we found increased nitrite production and endothelial nitric oxide synthase activation (supplemental Figure 4), possibly due to increased H2O2.18 Increased nitric oxide reacts with mitochondrial O2•−, forming peroxynitrite, which may exacerbate mitochondria dysfunction. In summary, we have shown that in vivo SOD2V16A is a strong predictor of cardiovascular dysfunction in SCD patients; in vitro, we have demonstrated that SOD2V16A overassociates with complex IV of the respiratory chain, reducing complex activity and resulting in increased mitochondrial O2− and extracellular H2O2 production.
Mechanistically, 1 limitation remains unsolved: how does SOD2V16A with normal enzymatic activity and subcellular location result in lower complex IV activity? SOD2 is embedded within the mitochondria supercomplex I:III:IV, stabilizing and protecting the supercomplex from mitochondrial O2•− damage.19 In this supercomplex, complex IV requires 14 subunits to maintain its activity.20 Subunit 4 has redox-sensitive cysteine residues that have been proposed to regulate complex IV conformation and activity under oxidative stress.21 Elevated mitochondrial O2•− likely enhances H2O2 formation, leading to thiol oxidation and possible conformational changes in complex IV, reducing its activity. However, the mechanism driving the interaction between SOD2V16A and complex IV remains elusive. SOD2 acetylation has been shown to increase mitochondrial O2•− production22; however, we found no increase in SOD2V16A acetylation at baseline or after treatment with antimycin A (supplemental Figure 5). Future studies examining endogenous SOD2V16A in endothelial cells and other cell types like platelets from SCD patients are needed.4
This study may better inform SCD patient management by using SOD2V16A as a genetic biomarker to determine efficacy to therapeutics and may help guide precision medicine approaches for SCD, particularly those therapies that reduce mitochondrial O2−.23 Current antioxidant therapies such as l-glutamine24 and hydroxyurea25 may be more effective in patients carrying the SOD2V16A polymorphism.
Acknowledgments
The authors thank Mark Gladwin for his insightful discussion regarding the data and methods. This study was supported, in part, by the American Society of Hematology (A.D.-O.). Schematics generated on biorender.com.
Financial support for this work was provided by the National Institutes of Health (NIH) (grants R01 HL 133864, R01 HL 128304, R01 HL153532, and R35 HL161177), American Heart Association (AHA) Established Investigator Award (19EIA34770095), and The Hemophilia Center of Western Pennsylvania, and Vitalant (A.C.S.).
Authorship
Contribution: A.D.-O. and A.C.S. designed the research questions, analyzed and interpreted data, and wrote the manuscript; S.Y. performed experiments, interpreted data, and assisted in writing the manuscript; M.R., S. Sanker, D.B.S., M.S., S.M.N., Y.Z., and L.G. performed experiments and analyzed and interpreted data; K.C.W. assisted in writing the paper; and B.A.K. and S. Shiva performed data analysis and data interpretation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Adam C. Straub, Department of Pharmacology and Chemical Biology, The Heart, Lung, Blood and Vascular Medicine Institute, University of Pittsburgh School of Medicine, E1254 Biomedical Science Tower, 200 Lothrop St, Pittsburgh, PA 15216; e-mail: astraub@pitt.edu.
Requests for data sharing may be submitted to Adam C. Straub (astraub@pitt.edu).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal