

TO THE EDITOR:
Clonal hematopoiesis (CH), defined by the acquisition of somatic mutations in hematopoietic stem cells, occurs in 20% to 30% of individuals >60 years and most frequently affects epigenetic regulator genes (DNMT3A, TET2, and ASXL1). CH is associated with a higher overall mortality and an approximately 10-fold risk for the development of hematologic malignancies.1,2 Reduced overall survival (OS) in individuals with CH is mainly caused by an increased rate of cardiovascular events.2,3 A causal relation was found in preclinical models, showing accelerated development of atherosclerosis driven by an altered function of the NLRP3/IL1β inflammasome of mutated monocytes/macrophages.3-5 These results pinpoint toward pleiotropic effects of mutated clones, not only affecting self-renewal and differentiation of hematopoietic stem cells but also inflammatory signaling of mature blood cells.
In patients with cancer, CH was reported to be associated with adverse outcomes such as a higher rate of therapy-associated myeloid neoplasia and reduced OS.6,7 However, current knowledge mainly derives from retrospective patient cohorts with marked heterogeneity in terms of cancer entity, disease stage, and applied therapy. Keeping unexpected findings associated with CH in mind, caution is needed to predict clinical consequences of CH to consider this phenomenon in all its bearings. Thus, we investigated CH in 237 available peripheral blood (PB) samples from the FIRE-3 study (AIO KRK-0306), a phase 3 landmark trial comparing FOLFIRI plus cetuximab or bevacizumab as first-line treatment in metastatic colorectal cancer (mCRC).8
In accordance with the Declaration of Helsinki and with approval of the local ethics committee of the University of Munich, PB samples collected as part of the screening process were available from 237 patients with mCRC of the FIRE-3 intention-to-treat (ITT) population.8 With respect to demographic and clinical characteristics, our subcohort is representative of the entire FIRE-3 ITT cohort (supplemental Table 1, available on the Blood Web site). PB DNA was subjected to error-corrected targeted sequencing using a customized sequencing panel covering 45 genes recurrently mutated in CH (supplemental Table 2). Somatic variants with a variant allele frequency (VAF) ≥ 1% were called using our in-house variant calling pipeline (detailed in supplemental Methods).9,10 The primary end point of our study was OS, and secondary end points included the occurrence of adverse events and various response measurements such as early tumor shrinkage. Statistical analysis was performed in R version 4.0.1 as described in the supplemental Material. Because of the hypothesis-generating character of the study, no correction for multiple testing was implemented, and reported P values must be interpreted as exploratory.
We identified 119 mutations in 86 patients, corresponding to a CH prevalence of 36%. The most frequently mutated genes were DNMT3A, TET2, PPM1D, and ASXL1, accounting for 72% of all mutations (Figure 1A). In 24 (10%) patients, more than 1 mutation was detected (Figure 1B). Comparisons of baseline demographic and clinical characteristics according to CH status confirmed the age-related association of CH as reported previously (median age: 62 vs 68 years in CHnegative vs CHpositive patients, respectively; P < .001; supplemental Table 3; Figure 1C). Baseline blood counts did not differ between groups (supplemental Figure 1). Prior exposure to chemotherapy was associated with a higher CH mutation rate (50% vs 33%; P = .04; Figure 1D). Genes related to the DNA damage repair machinery such as TP53, PPM1D, CHEK2, RAD21, BRCC3, and ATM were particularly affected (24% vs 8%; P = .006; supplemental Figure 2).
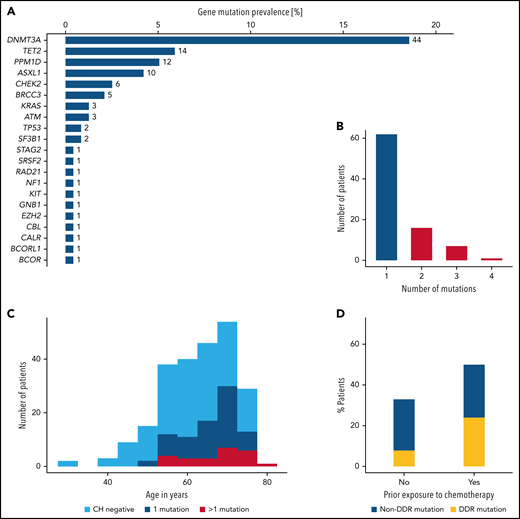
Overview of mutations identified by targeted sequencing. (A) Gene-specific prevalence of CH mutations. (B) Number of patients with 1, 2, 3, or 4 mutations. (C) Age distribution of 237 patients according to CH status. (D) CH prevalence according to prior exposure to chemotherapy. DDR, DNA-damage repair machinery.
Overview of mutations identified by targeted sequencing. (A) Gene-specific prevalence of CH mutations. (B) Number of patients with 1, 2, 3, or 4 mutations. (C) Age distribution of 237 patients according to CH status. (D) CH prevalence according to prior exposure to chemotherapy. DDR, DNA-damage repair machinery.
Next, we observed a longer but not statistically significant OS for patients who were CHpositive (median OS: 43.7 vs 33.2 months in patients who were CHnegative; Figure 2A). Because of the unbalanced age distribution, we presumed patient age to be a relevant confounder of the OS estimate. In a Cox proportional hazards model including age, treatment arm, liver-limited disease, synchrone vs metachrone metastasis, and Eastern Cooperative Oncology Group (ECOG) performance status as covariates, CH was an independent predictor of OS (hazard ratio [HR], 0.64; 95% confidence interval [CI], 0.46-0.89; P = .007; Figure 2C). This association was even more pronounced for patients with multiple mutations (HR, 0.37; 95% CI, 0.21-0.64; P < .001; supplemental Figure 3). In an exploratory analysis on single gene and gene group level for most frequently mutated genes, particularly DNMT3A-mutated patients (CH-DNMT3A) showed longer OS compared with DNMT3A wild-type patients: median OS was 51.1 vs 33.2 months (P = .044 in log-rank test; Figure 2B; supplemental Figure 4). CH-DNMT3A remained an independent predictor of OS in multivariate analysis (HR, 0.53; 95% CI, 0.36-0.80; P = .002; Figure 2D). In line with this, patients with CH-DNMT3A more frequently showed early tumor shrinkage,11 defined by a reduction of a reference tumor lesion of at least 20% after 6 or 8 weeks of treatment (75% vs 56%; P = .033; supplemental Table 4). Of note, the association with improved OS also holds for the more conservative definitions of clonal hematopoiesis of indeterminate potential12 (n = 65), with VAF ≥ 2%, and clonal hematopoiesis with potential driver mutations (n = 65) as defined in Coombs et al6 (supplemental Figures 5 and 6). In fact, VAF as a continuous measure of clone size is significantly associated with improved OS, hinting toward a potential dose-response relationship (supplemental Figure 7).
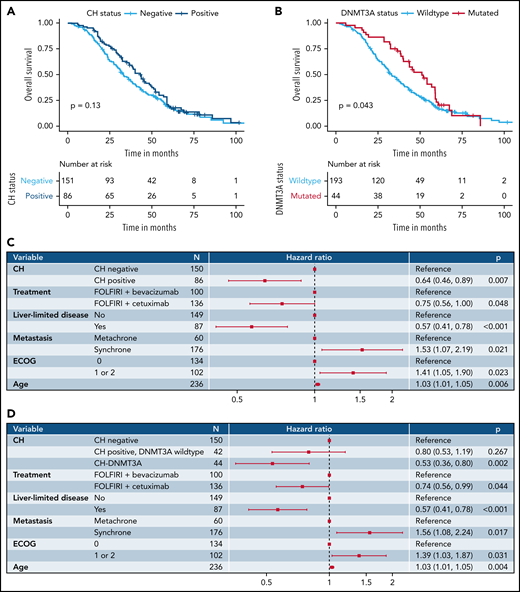
Impact of clonal hematopoiesis on clinical outcome. (A) Kaplan-Meier analysis of OS stratified by CH status in the FIRE-3 cohort. (B) Kaplan-Meier analysis of OS stratified by DNMT3A mutation status in the FIRE-3 cohort. (C) Cox proportional hazard model of OS with CH status, treatment arm, liver-limited disease status, history of metastasis, ECOG performance status, and age as covariates. (D) Cox proportional hazard model of OS with CH-DNMT3A status, treatment arm, liver-limited disease status, history of metastasis, ECOG performance status, and age as covariates.
Impact of clonal hematopoiesis on clinical outcome. (A) Kaplan-Meier analysis of OS stratified by CH status in the FIRE-3 cohort. (B) Kaplan-Meier analysis of OS stratified by DNMT3A mutation status in the FIRE-3 cohort. (C) Cox proportional hazard model of OS with CH status, treatment arm, liver-limited disease status, history of metastasis, ECOG performance status, and age as covariates. (D) Cox proportional hazard model of OS with CH-DNMT3A status, treatment arm, liver-limited disease status, history of metastasis, ECOG performance status, and age as covariates.
Regarding secondary end points in the FIRE-3 cohort, no association of CH with progression-free survival in first-line or subsequent treatment was found (supplemental Figure 8). Although there was no difference in time on first-line treatment, time on subsequent treatment lines was significantly longer for CH and CH-DNMT3A (supplemental Table 5). CH was associated with the occurrence of grade 3/4 diarrhea (17.4% vs 6.6%; P = .014) and the occurrence of thromboembolic events (7.0% vs 1.3%; P = .028; supplemental Tables 6 and 7; supplemental Figure 9). Importantly, neither association of CH with hematotoxic therapy complications nor with tumor mutational status13 nor with consensus molecular subgroups based on gene expression signatures14 was found (supplemental Tables 8 and 9).
To our knowledge, this is the first study of CH in a well-defined cohort of patients with cancer with prospectively collected clinical data. Surprisingly, CH and, in particular, CH-DNMT3A were associated with improved OS in this patient collective. Coombs et al6 reported shorter OS in 5650 patients with cancer, encompassing 51 cancer types with varying disease stages, treatment regimens, and blood sampling time points, making general conclusions for one cancer entity difficult.
Given the important role of CH in inflammatory processes, it is conceivable that CH mutations and, in particular, DNMT3A mutations in mature immune cells modulate the tumor microenvironment. Increased activity of the NLRP3 inflammasome has been linked to DNMT3A deficiency15 and was shown to play a role in the suppression of metastatic growth via interleukin-18 and activation of natural killer cells.16 Moreover, the polarization of protumorigenic and antitumorigenic phenotypes of tumor-infiltrating leukocytes (TILs) is in part controlled by epigenetic mechanisms.17 In fact, DNMT3A regulates the polarization of TH1 and TH2 cells via silencing of the interferon γ promoter.18,19 Along these lines, we hypothesize that DNTM3A mutations shift the polarization of TILs toward the antitumorigenic phenotype, leading to improved tumor control and OS.
Whether the observed association is tumor entity specific, therapy specific, or an independent factor in this context remains to be investigated in future mechanistic studies. In addition, confirmatory studies in large independent mCRC datasets are warranted in the future.
Acknowledgments
The authors thank all participating patients and their families of the FIRE-3 trial, the AIO KRK study group, and all participating centers. The authors also thank Tatiana Borodina and Jeannine Wilde for technical assistance and acknowledge the assistance of the Genomics Core Facility Berlin Institute of Health/Max Delbrück Center for Molecular Medicine.
FIRE-3 was financially supported by Merck KGaA and Pfizer. This study was supported by grants from the Deutsche Krebshilfe (70113643), and the Deutsches Konsortium für Translationale Krebsforschung awarded to F.D. C.M.A. received support from the Berlin Institute of Health junior clinical scientist program. S.D. was supported by a stipend from the Berlin School of Integrative Oncology. P.M.S. was supported by a postdoctoral research fellowship from the Alexander von Humboldt Foundation.
Funding support for this article was provided by the German Cancer Aid (70113643).
Authorship
Contribution: C.M.A. and F.D. designed the study; C.M.A., S.D., A.S., R.H., P.M.S., C.M.S., M.T., D.P.M., S.S., V.H., and F.D. provided cases, data, and/or clinical annotation; C.M.A., S.D., A.S., R.H., P.M.S., C.M.S., M.T., and F.D. carried out experiments and data analysis; C.M.A., S.D., V.H., and F.D. wrote the manuscript; and all authors reviewed and approved the final manuscript.
Conflict-of-interest disclosure: F.D. reports personal fees from Roche, Novartis, AbbVie, Incyte, and Astra Zeneca outside the submitted work. A.S. received honoraria by Roche, Taiho Pharmaceutical, and Servier and expenses for travel and accommodations by Roche, Merck KGaA, Amgen, Pfizer, and Lilly Oncology. L.B. reports personal fees from Abbvie, Amgen, Astellas, Bristol-Myers Squibb, Celgene, Daiichi Sankyo, Gilead, Hexal, Janssen, Jazz Pharmaceuticals, Menarini, Novartis, Pfizer, Sanofi, and Seattle Genetics outside the submitted work. D.P.M. reports honoraria and advisory roles from Amgen, Merck, Sanofi, Pfizer, BMS, MSD, Servier, Lilly, Taiho, Pierre Fabre, COR2ED, G1, EPICS, Onkowissen.de, and Incyte outside the submitted work. D.P.M. reports research grants (institutional) from Amgen and Servier. S.S. reports personal fees from Amgen, Bayer, Bristol-Myers Squibb, Daiichi Sankyo; Esai, Lilly, Merck KGaA, MSD, Pierre Fabre, Roche, Sanofi, Taiho, and Takeda outside the submitted work. All remaining authors declare no competing financial interests.
Correspondence: Frederik Damm, Department of Hematology, Oncology, and Cancer Immunology, Charité Universitätsmedizin Berlin, Augustenburger Platz 1, 13353 Berlin, Germany; e-mail: frederik.damm@charite.de.
Detailed information regarding the gene panel and DNA sequencing is available from the corresponding author upon reasonable request.
The online version of this article contains a data supplement.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal