

In this issue of Blood, uncover a key role that programmed death ligand 1 (PD-L1) plays in delaying neutrophil apoptosis at the site of inflammation by activation of the phosphatidylinositol 3-kinase (PI3K)-AKT survival pathway.1
Polymorphonuclear neutrophils (PMNs, or neutrophils) are the most abundant circulating leukocytes, constituting 60% to 70% of circulating white blood cells. PMNs are terminally differentiated and have a short lifespan, but are essential for innate immunity and host defense against microbes.2 They are the first cells to be massively recruited at the site of infection where they recognize microbes via different receptors, inducing engulfment of the microbe into a phagosome.3,4 Killing of microbes by PMNs occurs through the release, into the phagosome, of toxic agents such as reactive oxygen species and the content of granules (myeloperoxidase, glucosidases, proteases, and antibacterial peptides, etc). Microbes can also be trapped and killed by neutrophil extracellular traps (NETs).5 Many processes such as apoptosis, NETosis, pyroptosis, and necroptosis can induce the death of neutrophils,5 upon which they are phagocytized and eliminated by macrophages through a process called efferocytosis, resulting in the cleaning of the infection site. Thus, PMNs are critical anti-inflammatory components of the innate immune system as their physiological role is to resolve both infection and inflammation. Nevertheless, excessively activated or delayed apoptosis results in PMNs becoming harmful to surrounding tissues due to cell injury and continued inflammatory reaction, the driving factors for inflammatory disorders such as sepsis.4
Tissue neutrophils are believed to have longer lifespans than circulating PMNs, due to the presence of survival factors at the inflammatory site. Extended neutrophil lifespan through apoptosis suppression has been reported in patients with several inflammatory diseases and is associated with increased disease severity.6 Neutrophils isolated from blood die through constitutive apoptosis, which can be either accelerated or inhibited by several agents. Inhibition of neutrophil apoptosis can contribute to inflammation; however, the factors leading to dysregulation of neutrophil apoptosis in inflammation are still not completely identified. In this issue, Wang et al demonstrate that PD-L1 plays a key role by delaying neutrophil apoptosis at the site of inflammation through the PI3K-AKT pathway (see figure).
Programmed cell death 1 (PD-1) protein, expressed on immune cells, is an immune checkpoint inhibitory receptor that triggers immunosuppressive signaling pathways.7 PD-1 binds to PD-L1 or PD-L2 and blocks activating signals from T-cell receptors. PD-1/PD-L1 function as brakes to limit the adaptive immune response and the beneficial T-cell functions in cancer. PD-L1 is expressed on the plasma membrane of T and B cells and antigen-presenting cells.7 Cancer cells also express PD-L1, which binds to the T-cell surface via PD-1 allowing them to escape host immune response. Thus, anti-PD-1/anti-PD-L1 antibodies have been used to treat various types of cancer. Previous studies have shown that PD-L1 expression on neutrophils increases in various inflammatory conditions.8,9 In this new study, Wang et al show that PD-L1 overexpression in neutrophils from septic patients correlates with neutrophil survival. Silencing PD-L1 expression using small interfering RNA (siRNA) accelerated apoptosis of septic neutrophils. Interestingly, neutrophils challenged with interferon γ (IFN-γ) plus lipopolysaccharide (LPS) and neutrophils from septic patients exhibited increased AKT phosphorylation, which was reversed by PD-L1 siRNA. The authors found that PD-L1 coimmunoprecipitates with the p85 subunit of PI3K, the upstream regulator of AKT. In vivo, neutrophil PD-L1 deletion reduced neutrophil lung infiltration in a cecal ligation and puncture murine model and attenuated lung injury. Thus, increased PD-L1 expression on human neutrophils during inflammation delays cellular apoptosis via the PI3K-AKT pathway, driving lung injury and mortality.
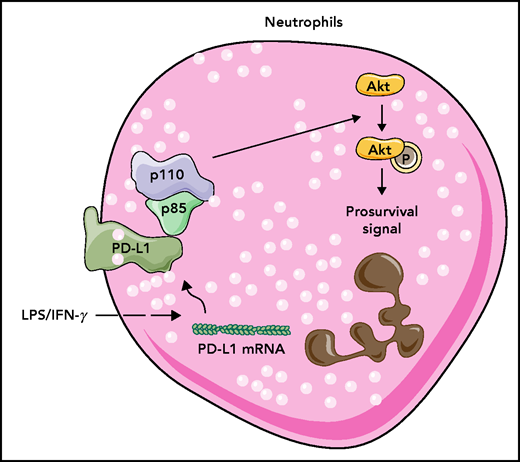
LPS plus IFN-γ and a septic environment induce PD-L1 expression in human neutrophils. PD-L1 binds to p85, the PI3K regulatory subunit, and PI3K activates AKT kinase to induce prosurvival signals in neutrophils making them live longer. mRNA, messenger RNA; p (p110 and p85), protein; P (Akt), phosphorylation.
LPS plus IFN-γ and a septic environment induce PD-L1 expression in human neutrophils. PD-L1 binds to p85, the PI3K regulatory subunit, and PI3K activates AKT kinase to induce prosurvival signals in neutrophils making them live longer. mRNA, messenger RNA; p (p110 and p85), protein; P (Akt), phosphorylation.
The study by Wang et al brings significant new information to the neutrophil field. The first important message is that PD-L1 is not only a key player in cancer immunomodulation, but also in inflammation, raising the possibility that it could be a novel pharmacological target. The second message is that PD-L1 upregulation in neutrophils during sepsis promotes their survival. Finally, this study highlights the novel and important role of PD-L1 as an inducer of the PI3K-AKT signaling pathway, which regulates many neutrophil inflammatory functions. Thus, this study adds another critical step to our understanding of neutrophil biology.
As is always the case with novel mechanisms, several questions have been raised. In cancer, PD-L1 is expressed on the plasma membrane, but its localization in neutrophils is not addressed by the authors. Knowing whether PD-L1 is expressed on the plasma membrane or on the membrane of neutrophil granules would be interesting. Expression of PD-L1 on the plasma membrane would make it an easy target for immunotherapies with blocking antibodies. Indeed, while this study was under revision, Thanabalasuriar et al10 reported the role of PD-L1+ neutrophils in airway inflammation in mice and showed that an anti-PD-L1 antibody protected mice from inflammation. Patera et al9 have also shown that inflammatory neutrophils express PD-1; the intercellular PD-1/PD-L1 engagement on PMNs could induce PI3K-AKT activation in neutrophils. This possibility could be checked using blocking antibodies. Wang et al showed that IFN-γ plus LPS induced PD-L1 expression in neutrophils; however, it would be of interest to know whether other inflammatory stimuli also induce PD-L1 expression in PMNs and by which mechanisms.
Although this study suggests a fundamental role of the PI3K-AKT pathway in mediating PD-L1–induced neutrophil survival, the mechanism by which PD-L1 activates PI3K was not investigated as the authors only show its ability to bind p85 when expressed in HEK293 cells. This pathway should be checked in inflammatory neutrophils. In addition, the PI3K-AKT pathway could modulate several other important neutrophil functions including superoxide production, degranulation, and chemotaxis, raising the question of whether these functions are also impacted in PD-L1+ neutrophils. Furthermore, evaluating the role of PD-L1 in other inflammatory diseases such as rheumatoid arthritis and inflammatory bowel diseases would be of interest.
In summary, the study by Wang et al represents a major advance in our understanding of neutrophil biology. Notably, their work uncovers the important function of the PD-L1–PI3K–AKT axis in delaying neutrophil apoptosis in inflammatory situations, opening new avenues for novel therapeutic approaches in several immunological and inflammatory diseases and raising intriguing questions about PD-L1 to be explored in the future.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal