

Key Points
Nonspecific radiographic and CSF findings result in delayed diagnosis and treatment when PCNSL is suspected.
Integrating a rapid genotyping assay into workup of PCNSL can potentially resolve treatment delays and obviate neurosurgical sampling.

Abstract
Diagnosing primary central nervous system lymphoma (PCNSL) frequently requires neurosurgical biopsy due to nonspecific radiologic features and the low yield of cerebrospinal fluid (CSF) studies. We characterized the clinical evaluation of suspected PCNSL (N = 1007 patients) and designed a rapid multiplexed genotyping assay for MYD88, TERT promoter, IDH1/2, H3F3A, and BRAF mutations to facilitate the diagnosis of PCNSL from CSF and detect other neoplasms in the differential diagnosis. Among 159 patients with confirmed PCNSL, the median time to secure a diagnosis of PCNSL was 10 days, with a range of 0 to 617 days. Permanent histopathology confirmed PCNSL in 142 of 152 biopsies (93.4%), whereas CSF analyses were diagnostic in only 15/113 samplings (13.3%). Among 86 archived clinical specimens, our targeted genotyping assay accurately detected hematologic malignancies with 57.6% sensitivity and 100% specificity (95% confidence interval [CI]: 44.1% to 70.4% and 87.2% to 100%, respectively). MYD88 and TERT promoter mutations were prospectively identified in DNA extracts of CSF obtained from patients with PCNSL and glioblastoma, respectively, within 80 minutes. Across 132 specimens, hallmark mutations indicating the presence of malignancy were detected with 65.8% sensitivity and 100% specificity (95% CI: 56.2%-74.5% and 83.9%-100%, respectively). This targeted genotyping approach offers a rapid, scalable adjunct to reduce diagnostic and treatment delays in PCNSL.
Introduction
Primary central nervous system lymphoma (PCNSL) is an aggressive extranodal non-Hodgkin lymphoma. Shorter time to diagnosis correlates with improved prognosis,1 whereas untreated PCNSL is associated with overall survival of <2 months.2 Distinguishing PCNSL from entities with similar radiographic appearance, including glioma, metastatic disease, or leptomeningeal carcinomatosis, remains challenging in clinical practice. The diagnostic workup of suspected PCNSL frequently involves analysis of cerebrospinal fluid (CSF) by cytopathology and flow cytometry, the former of which may be positive in leptomeningeal spread from other neoplasms.3 This diagnostic dilemma also complicates neurosurgical decision making; whereas the role of surgery in hematologic malignancies is limited to diagnostic biopsy,2 the initial diagnostic procedure for gliomas typically aims for maximal safe resection.4
Up to 70% of PCNSL, 24% to 73% of systemic diffuse large B-cell lymphomas, and 87% of Waldenström macroglobulinemia harbor the canonical MYD88 L265P mutation.5,6 Detection of this mutation in DNA from CSF in patients with PCNSL by digital droplet polymerase chain reaction7 raises the possibility that its detection may yield diagnostic information, but this approach cannot be performed for >1 to 2 genes simultaneously. Furthermore, the possibility of false negative results needs to be counterbalanced by positive detection of mutations commonly observed in other central nervous system (CNS) malignancies.8-12
In this study, we first detail the challenges in diagnosing PCNSL. To improve this workflow, we engineered a multiplexed quantitative polymerase chain reaction (qPCR)-based approach to identify canonical variants in PCNSL and glioma from tissue and liquid biopsies. Timely return of molecular diagnostic results foreshortens the times to PCNSL diagnosis and may obviate some neurosurgical biopsies.
Study design
Case records and specimens
CSF and peripheral blood specimens from patients with CNS pathologies were collected prospectively with informed consent under institutional review board approval at Massachusetts General Hospital (MGH) (supplemental Methods, available on the Blood Web site; supplemental Figure 1). Retrospective case records review and archived tumor specimen collection were performed under institutional review board approval at MGH, Dana-Farber Cancer Institute, and Brigham and Women’s Hospital.
Genotyping assay
We engineered qPCR-based assays for detecting mutations found in PCNSL and glioma, including MYD88 L265P, TERT promoter, IDH1/2, H3F3A, and BRAF mutations,11 further adapted for speed and sensitivity from CSF and plasma (supplemental Methods).
Statistics
Diagnosis, treatment, follow-up, and survival were calculated with respect to the date of admission to our facility. Sensitivity and specificity thresholds were calculated by the Clopper-Pearson method.
Results and discussion
We analyzed the clinical courses of 159 consecutive patients diagnosed with PCNSL between 2010 and 2019, in order to characterize the existing diagnostic workflow for this entity. PCNSL was not included as a potential diagnosis in the initial evaluation of 27 patients (17%) (supplemental Table 1). Median time to diagnosis was 10 days (range, 0-617; interquartile range, 7-17 days) (Figure 1A). Patients initially undergoing CSF sampling required 1 to 5 additional procedures prior to establishing a diagnosis (supplemental Figure 2A).
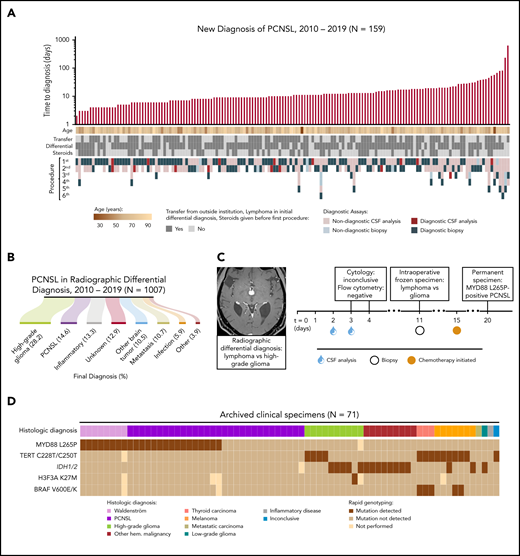
The TetRS qPCR-based rapid genotyping panel detects recurrent molecular alterations relevant to the diverse radiologic differential diagnosis of PCNSL and subsequent diagnostic workup. (A) Clinical characteristics of 159 patients who established a new diagnosis of PCNSL at our facility. Diagnostic assays include all procedures performed at our facility and outside facilities. Time to diagnosis is displayed on a logarithmic scale, calculated from date of admission to our facility. One patient is not displayed, who had 4 lumbar punctures as an outpatient, with the final assay returning positive on the first day of inpatient admission. This cohort included 2 HIV-positive (1.3%), 17 EBV-positive (10.7%), and 13 immunosuppressed patients (8.2%) (supplemental Table 1). (B) Final diagnosis among patients with a new brain lesion with PCNSL in the differential diagnosis. (C) A representative patient with a new brain lesion underwent 2 nondiagnostic lumbar punctures prior to a brain biopsy. Intraoperative histopathology was inconclusive. Chemotherapy was initiated empirically prior to final pathologic diagnosis due to symptomatic decline. Analysis of archived biopsy tissue revealed the MYD88 L265P mutation. (D) Results of TetRS assay on DNA extracts from 71 archived, clinically annotated biopsy specimens by sensitive detection of hotspot mutations in MYD88, TERT promoter, IDH1/2, H3F3A, and BRAF. Expanded results are shown in supplemental Figure 7. EBV, Epstein-Barr virus; Hem., hematologic; HIV, human immunodeficiency virus.
The TetRS qPCR-based rapid genotyping panel detects recurrent molecular alterations relevant to the diverse radiologic differential diagnosis of PCNSL and subsequent diagnostic workup. (A) Clinical characteristics of 159 patients who established a new diagnosis of PCNSL at our facility. Diagnostic assays include all procedures performed at our facility and outside facilities. Time to diagnosis is displayed on a logarithmic scale, calculated from date of admission to our facility. One patient is not displayed, who had 4 lumbar punctures as an outpatient, with the final assay returning positive on the first day of inpatient admission. This cohort included 2 HIV-positive (1.3%), 17 EBV-positive (10.7%), and 13 immunosuppressed patients (8.2%) (supplemental Table 1). (B) Final diagnosis among patients with a new brain lesion with PCNSL in the differential diagnosis. (C) A representative patient with a new brain lesion underwent 2 nondiagnostic lumbar punctures prior to a brain biopsy. Intraoperative histopathology was inconclusive. Chemotherapy was initiated empirically prior to final pathologic diagnosis due to symptomatic decline. Analysis of archived biopsy tissue revealed the MYD88 L265P mutation. (D) Results of TetRS assay on DNA extracts from 71 archived, clinically annotated biopsy specimens by sensitive detection of hotspot mutations in MYD88, TERT promoter, IDH1/2, H3F3A, and BRAF. Expanded results are shown in supplemental Figure 7. EBV, Epstein-Barr virus; Hem., hematologic; HIV, human immunodeficiency virus.
Although permanent histopathologic interpretation was eventually diagnostic of PCNSL in 93.4% of initial biopsies, CSF analyses (including cytopathology, flow cytometry, and/or immunoglobulin H [IgH] gene rearrangement) were diagnostic in only 13.3% of studies (supplemental Tables 1 and 2). Final diagnoses were secured by biopsy (148/159 patients, 93.1%), CSF (6/159, 3.8%), or radiographic grounds alone (5/159, 3.1%) (supplemental Table 3). All diagnoses secured by CSF analysis had diagnostic flow cytometry and/or IgH rearrangement; none were secured by cytopathology alone. Delays in IgH rearrangement results led to 5 patients undergoing biopsy (supplemental Table 4).
Indicative of the low sensitivity and delays associated with CSF sampling (performed in 69.8% of patients), ultimately establishing PCNSL diagnosis required neurosurgical biopsy in 95.64% of cases by craniotomy (41.7%) or stereotactic needle (58.3%) (supplemental Table 2). Postoperative complications were noted with biopsies (supplemental Table 5). Furthermore, intraoperative histopathologic analysis confirmed PCNSL in 45.5% of cases, whereas the diagnosis of lymphoma was inconclusive (12.2%) or not raised (42.3%) in the remainder (supplemental Table 4). The inability to secure a diagnosis of PCNSL intraoperatively likely contributed to performing surgical resection in 35 cases (22.2%) (supplemental Table 2).
To place this evaluation of patients ultimately diagnosed with PCNSL in the context of nonspecific imaging findings, we examined 1007 patients with a new brain lesion that included PCNSL in the radiologic differential diagnosis. The most common final diagnoses were high-grade glioma (28.2%) and PCNSL (14.6%), with the remainder yielding diverse neoplastic, inflammatory, and infectious etiologies (Figure 1B). An illustrative case of diagnostic delay in PCNSL is highlighted by a patient with inconclusive results from 2 CSF samplings, and intraoperative histology suggestive of either lymphoma or glioma. Pathologic diagnosis of PCNSL was secured on postadmission day 20, with subsequent detection of the MYD88 L265P mutation (Figure 1C).
Noting diagnostic delays and risks associated with invasive procedures in such cases with difficult to access lesions, we engineered our previously described qPCR method for rapid genotyping11 to detect canonical mutations, including MYD88 L265P with a lower limit of 0.15% mutant allele fraction (supplemental Figures 2-6). We were able to distinguish PCNSL from glioma by combining MYD88 mutation detection with parallel analysis of TERT promoter, IDH1/2, H3F3A, and BRAF point mutations. Results of our platform, termed targeted rapid sequencing (TetRS), were concordant with histologic diagnoses and orthogonal DNA sequencing in clinical tumor specimens and patient-derived cell line extracts representing diverse CNS malignances (Figure 1D). Across 86 archived specimens, including 34 MYD88 L265P mutant tumors, we detected this variant with 100% sensitivity and specificity (95% confidence interval [CI]: 89.7-100 and 93.2-100, respectively; supplemental Figure 8). Detection of MYD88 L265P enabled identification of hematologic malignancies (including PCNSL, Waldenström macroglobulinemia, myelodysplastic syndrome, chronic myelogenous leukemia, acute myeloid leukemia, and secondary CNS lymphoma) with 57.6% sensitivity and 100% specificity (95% CI: 44.1-70.4 and 87.2-100, respectively; supplemental Figure 8).
TetRS was then optimized to report results from CSF and plasma specimens within 80 minutes (supplemental Figures 9-10) and prospectively validated on liquid biopsies from 32 patients (Figure 2A; supplemental Figures 11-12). We represent 2 patients with positive TetRS findings in CSF. The MYD88 L265P mutation was detected in CSF from a patient who underwent suboccipital craniectomy for gross total resection of PCNSL. In another case, the TERT C228T mutation was detected in CSF from a patient with suspected PCNSL, but ultimately diagnosed with glioblastoma following brain biopsy (Figure 2B-C). Of note, analysis of CSF from a patient presenting with leptomeningeal disease secondary to systemic diffuse large B-cell lymphoma revealed TERT C228T and MYD88 L265P comutation (Figure 2A), an alteration that has been previously reported in CNS lymphoma.13 Detection of MYD88 L265P in CSF by TetRS was 100% concordant with available clinical molecular testing (supplemental Table 8). In 132 specimens representing 117 patients, TetRS detected hallmark mutations of malignancies with 65.8% sensitivity and 100% specificity (95% CI: 56.2-74.5 and 83.9-100, respectively) (supplemental Figure 11).
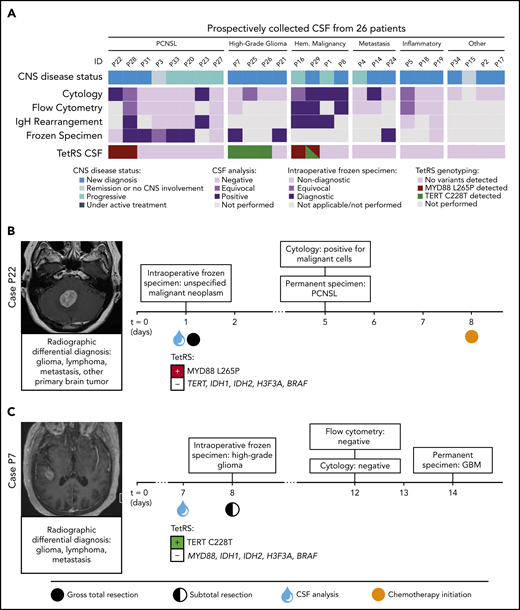
Detection of PCNSL- and glioma-specific somatic variants with the TetRS rapid genotyping assay in prospectively collected liquid biopsies. (A) The top row of segmented color bars displays the CNS disease status. The next 4 rows represent the results of routine clinical CSF studies and intraoperative frozen histopathology. The bottom row shows the mutant alleles detected in CSF by the TetRS rapid genotyping assay. Expanded results are shown in supplemental Figure 8. (B) Case P22 shows detection of the MYD88 L265P mutation in CSF from a patient with a cerebellar mass, who underwent gross total resection due to nondiagnostic intraoperative frozen specimen analysis. Cytology and permanent histologic analysis revealed PCNSL 4 days postoperatively. (C) Case P7 shows detection of the TERT C228T mutation in the CSF of a patient with a right temporal mass. Surgical needle biopsy was concluded after intraoperative frozen analysis showed high-grade glioma but converted to open craniotomy and subtotal resection due to intraoperative bleeding. GBM, glioblastoma multiforme; HGG, high-grade glioma.
Detection of PCNSL- and glioma-specific somatic variants with the TetRS rapid genotyping assay in prospectively collected liquid biopsies. (A) The top row of segmented color bars displays the CNS disease status. The next 4 rows represent the results of routine clinical CSF studies and intraoperative frozen histopathology. The bottom row shows the mutant alleles detected in CSF by the TetRS rapid genotyping assay. Expanded results are shown in supplemental Figure 8. (B) Case P22 shows detection of the MYD88 L265P mutation in CSF from a patient with a cerebellar mass, who underwent gross total resection due to nondiagnostic intraoperative frozen specimen analysis. Cytology and permanent histologic analysis revealed PCNSL 4 days postoperatively. (C) Case P7 shows detection of the TERT C228T mutation in the CSF of a patient with a right temporal mass. Surgical needle biopsy was concluded after intraoperative frozen analysis showed high-grade glioma but converted to open craniotomy and subtotal resection due to intraoperative bleeding. GBM, glioblastoma multiforme; HGG, high-grade glioma.
Minimizing false positive results is critical in the workup of low-prevalence diseases, such as PCNSL and CNS malignancies. TetRS is a technically straightforward approach to rapidly detect clinically relevant mutations in CNS malignancies when PCNSL is suspected. Variant alleles can be detected from solid or liquid biopsies within 37 or 63 to 80 minutes, respectively, with high specificity, threshold sensitivity as low as 0.15% to 1% mutant allele fraction,11 and reagent cost <$9.00 per specimen.
Integrating TetRS into the diagnostic workflow of suspected CNS malignancy has the potential to resolve delays in the clinical management of these patients (supplemental Figure 13). Optimal patient selection for invasive procedures could speed delivery of adjuvant therapies, such as radiation or chemotherapy, that could otherwise be delayed due to surgical recovery or diagnostic ambiguity. Targeted genotyping can guide cost-effective selection of next-generation sequencing panels, molecular assays,14 or assignment to clinical trials. Serial CSF sampling may also enable quantitative treatment response assessment.9 Because CNS lymphoma tumor DNA can be detected in CSF prior to radiographic recurrence,15 in future studies we will evaluate this method for tracking minimal residual or leptomeningeal disease, with the potential to guide decisions regarding salvage therapies prior to symptomatic progression.
Deidentified individual participant data underlying the reported results are available and proposals for access to this data should be sent to gshankar@mgh.harvard.edu.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the staff of the Massachussetts General Hospital/Brigham and Women's Hospital (MGH/BWH) Department of Pathology tissue banks for their guidance and support. They are deeply grateful for the partnership of the staff of the MGH Chemistry and Hematology clinical core laboratories, Brain Tumor Research Center, and Departments of Neuro-Radiology, Neurology, and Neuro-Oncology. This work would not have been possible without the tireless patient care and clinical research efforts of the MGH Neurology and Neuro-Oncology teams.
The funding agencies had no role in the design and conduct of the study, collection, management, analysis and interpretation of the data, preparation, review, or approval of the manuscript, or decision to submit the manuscript for publication.
This work was supported by grants from the National Institutes of Health (NIH) National Institute of Neurological Disorders and Stroke 1K08NS107634-01A1 (G.M.S.), as well as from the NIH National Cancer Institute 5U01CA230697 (B.S.C.).
Authorship
Contribution: G.M.S., B.S.C., D.P.C., J.D., J.M.R., J.K.L., H.D.M., and P.K.B. conceived of and designed the study; M.G., E.J.B., N.Z.G., J.T., N.N., K.T., and G.M.S. acquired, analyzed, and interpreted data; M.G. and G.M.S. performed statistical analysis; G.M.S., B.S.C., D.P.C., J.D., K.T., H.D.M., P.K.B., J.M.R., and J.K.L. provided administrative, technical and material support; G.M.S., B.S.C., D.P.C., and J.D. supervised the study; G.M.S. had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis; and all authors participated in drafting and critically revising the manuscript for important intellectual content.
Conflict-of-interest disclosure: P.K.B. has consulted for Angiochem, Genentech-Roche, Lilly, Tesaro, ElevateBio, Pfizer (Array), SK Life Sciences, and Dantari, received grant/research support (to MGH) from Merck, BMS, and Lilly, and received honoraria from Merck, Genentech-Roche, and Lilly. M.J.F. has advisory roles in Celgene/BMS, Takeda, Novartis, Kite/Gilead, and Arcellx. Outside the scope of the current work, D.P.C. has consulted for Lilly and Boston Pharmaceuticals and has received honoraria and travel reimbursement from Merck for invited lectures as well as from the NIH and US Department of Defense for clinical trial and grant review. B.V.N. has consulted for Robeaute and BK Medical and served as a cofounder and consultant for REACT Neuro. J.T.J. receives royalties from Elsevier; has consulted for CereXis, Health2047, and NF Network; and has consulted for and holds <1% stock in Navio Theragnostics. J.D. is a consultant for Unum Therapeutics, Blue Earth Diagnostics, and Magnolia; has received royalties from Wolters Kluwer; and has received research support from Beacon Biosignals, Boehringer Ingelheim, Brystol-Myers Squibb, Medimmune, Acerta Pharma, Orbus Therapeutics, and Novartis Pharmaceuticals. T.T.B. serves on the scientific advisory board for Genomicare; receives royalties from UpToDate and Oxford University Press; and receives honoraria from Oakstone Publishing and Audiodigest. The remaining authors declare no competing financial interests.
Correspondence: Ganesh M. Shankar, Department of Neurosurgery, Massachusetts General Hospital, 15 Parkman St, WAC 745, Boston, MA 02114; e-mail: gshankar@mgh.harvard.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal