


Abstract
Venetoclax-based regimens have expanded the therapeutic options for patients with chronic lymphocytic leukemia (CLL), frequently achieving remissions with undetectable measurable residual disease and facilitating time-limited treatment without chemotherapy. Although response rates are high and durable disease control is common, longer-term follow-up of patients with relapsed and refractory disease, especially in the presence of TP53 aberrations, demonstrates frequent disease resistance and progression. Although the understanding of venetoclax resistance remains incomplete, progressive disease is typified by oligoclonal leukemic populations with distinct resistance mechanisms, including BCL2 mutations, upregulation of alternative BCL2 family proteins, and genomic instability. Although most commonly observed in heavily pretreated patients with disease refractory to fludarabine and harboring complex karyotype, Richter transformation presents a distinct and challenging manifestation of venetoclax resistance. For patients with progressive CLL after venetoclax, treatment options include B-cell receptor pathway inhibitors, allogeneic stem cell transplantation, chimeric antigen receptor T cells, and venetoclax retreatment for those with disease relapsing after time-limited therapy. However, data to inform clinical decisions for these patients are limited. We review the biology of venetoclax resistance and outline an approach to the common clinical scenarios encountered after venetoclax-based therapy that will increasingly confront practicing clinicians.
Introduction
The BCL2 inhibitor (BCL2i), venetoclax, and Bruton tyrosine kinase inhibitors (BTKi's) have significantly improved outcomes for patients with CLL, especially those whose disease harbors TP53 aberrations or unmutated immunoglobulin heavy chain variable region (IGHV) status.1-5 Venetoclax-based regimens frequently achieve undetectable measurable residual disease (uMRD; <1 CLL cell per 104 leukocytes) in the peripheral blood (PB) and bone marrow (BM), which is strongly associated with prolonged progression-free survival (PFS).5-9 Attainment of deep remissions facilitates drug withdrawal and time-limited treatment strategies, which challenge the paradigm of indefinite targeted agent monotherapy. In the MURANO study, 24 months of venetoclax combined with 6 doses of monthly rituximab achieved superior PFS and overall survival (OS), compared with bendamustine-rituximab for patients with relapsed/refractory (RR) disease.4 For patients who received venetoclax-rituximab, 62% attained PB uMRD at the end of combination treatment (EOCT),10 the overall median PFS was 54 months, and 5-year OS was 82%.11 Among patients with end of treatment (EOT) PB uMRD, 39% maintained uMRD status 3 years after completing therapy, demonstrating that deep treatment-free remissions can be sustained after time-limited combination.11 The CLL14 trial randomized treatment-naive patients with comorbidities or renal impairment to 12 months of venetoclax or chlorambucil, each combined with 6 cycles of obinutuzumab, and demonstrated superior PFS after the venetoclax regimen with comparable toxicity.5 In the venetoclax-obinutuzumab cohort, 76% of patients attained PB uMRD12 and the 3-year PFS was 82%.13 Before these landmark trials, continuous venetoclax monotherapy had demonstrated efficacy in phase 2 studies of patients with disease harboring del(17p) (objective response rate [ORR], 77%; complete response rate [CRR], 20%)14 and progressive disease (PD) after B-cell receptor pathway inhibitors (BCRi's) (ORR, 65% to 67%; CRR, 5% to 9%)15,16 ; however, these subgroups are now included in the broader approvals for venetoclax-combinations after the MURANO and CLL14 trials. The advantages of time-limited venetoclax-combination regimens include frequent uMRD attainment, sustained treatment-free remissions, reduced cost and pill burden, and likely less cumulative toxicity compared with indefinite therapy.17-19 Potentially more significant, however, is the theoretical benefit of withdrawing selection pressure for venetoclax-resistant clones and the possibility of retreatment, given that BCL2 resistance mutations are typically observed after prolonged drug exposure.20,21 Despite the efficacy of venetoclax-based regimens, MRD recrudescence and PD are common with extended follow-up.6,11 After venetoclax-rituximab in the MURANO trial, the 18-month PFS was 90%, 64%, and 8% for patients with EOT uMRD, low-positive MRD (10−4 –10−2), and high-positive MRD (>10−2), respectively.10 At latest follow-up, the median time toMRD recrudescence among patients who attained EOT uMRD was 19 months,with a median time to clinical PD of 25 months after MRD conversion.11 With expanding use ofvenetoclax, clinicians will increasingly encounter patients with PD after priorexposure. Herein, we outline the mechanisms of venetoclax resistance andour approach to patient management in a series of illustrative cases.
Mechanisms of venetoclax resistance
Clinicopathological associations with resistance
Current insights into venetoclax resistance are predominantly derived from patients treated with continuous venetoclax monotherapy for RR CLL, and their implications for patients receiving time-limited combination therapy are uncertain. On multivariate analysis of pooled early trial data, the clinicopathological factors independently associated with earlier progression were nodal bulk ≥5 cm, refractoriness to BCRis or fludarabine, TP53 aberrations (mutations and/or deletions), and NOTCH1 mutations.7 In the venetoclax-rituximab cohort of the MURANO study, genomic complexity assessed by high-density array comparative genomic hybridization was associated with inferior PFS, and TP53, NOTCH1, BRAF, and BIRC3 mutations were associated with lower EOT uMRD rates.22 Among patients attaining uMRD, del(17p), genomic complexity or unmutated IGHV status were associated with earlier MRD recrudescence and PD.11 On multivariate analysis of the venetoclax-obinutuzumab cohort of the CLL14 study, only del(17p) was independently associated with inferior PFS.23 The presence of complex karyotype (CK) by metaphase karyotyping was not significantly associated with inferior response depth or PFS; however, longer follow-up is needed to confirm this observation.24 Overall, TP53 aberrations do not compromise initial ORRs to venetoclax-based therapy,5,8,9 but are associated with inferior response depth and accelerated disease re-emergence, paralleling observations for patients receiving ibrutinib.25
Resistance biology
Intrinsic de novo venetoclax resistance is rare, even in high-risk disease, and combination therapy with anti-CD20 monoclonal antibodies achieves ORRs of 85% to 92%.4,5,8 Among patients treated with navitoclax, the BCL2/BCL-XL inhibitor which preceded venetoclax, clinical responses were inversely associated with increased expression of an alternative BCL2 family protein, MCL-1, and decreased expression of BIM, the proapoptotic protein whose activity is unleashed by BCL2 inhibition.26 Microenvironmental signals may upregulate BCL-XL, MCL-1, and BLF-1, attenuating reliance on BCL2 for cell survival. Preclinical data suggest that anti-CD20 monoclonal antibodies or BCRis may interrupt this mechanism and restore venetoclax sensitivity,27-30 consistent with high ORRs observed in combination trials.5,31
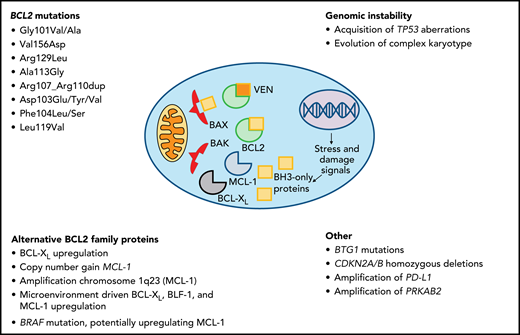
The best characterized mechanisms of acquired resistance include BCL2 mutations and upregulation of alternative prosurvival proteins. Acquisition of genomic instability and other mutations have been described as early events in small series, although their specific roles in venetoclax resistance require further investigation. BCL2 mutations are detected after sustained venetoclax exposure (median 36 months) and precede clinical PD by many months. The first identified mutation, BCL2 Gly101Val, compromises the binding affinity of venetoclax in the α-helical grove of BCL2 while preserving the protein’s antiapoptotic function, and has not been detected in pretreatment samples.21 Several other BCL2 resistance mutations have now been identified.20,32 Among 11 patients with venetoclax-resistant CLL harboring BCL2 Gly101Val, multiple distinct BCL2 mutations co-occurred in the disease of individual patients (median, 3; range, 1-7). Despite this, the proportion of CLL cells bearing any BCL2 mutation varied from <1% to 83%, implying alternative resistance mechanisms in the remaining leukemic population.20 In 1 patient, a distinct subclone lacking the BCL2 Gly101Val mutation overexpressed BCL-XL.21 In another case, a subclonal MCL-1 locus focal copy number gain was identified.20,21 At our centers, we have reported several instances of new TP53 aberrations or CK at progression on venetoclax, implicating genomic instability in the development of resistance.33,34 Other potential mechanisms include chromosome 1q23 amplification, with resultant overexpression of MCL-1 and changes to mitochondrial metabolism.35 Whole-exome sequencing of 8 patients with early PD (<24 months) and frequent Richter transformation (RT) identified recurrent mutations in BTG1 and homozygous deletions of CDKN2A/B. PD-L1 amplification was identified in 1 case, as was a BRAF mutation postulated to augment MCL-1 expression.36 These putative mechanisms of early resistance most likely differ from those driving PD after durable initial responses to venetoclax, which are typified by oligoclonal leukemic populations with multiple distinct resistance mechanisms, including frequent BCL2 mutations (Figure 1).
Case 1
A 74-year-old man was referred with progressive CLL after 2 prior chlorambucil-based regimens and fludarabine, cyclophosphamide, and rituximab (FCR). His disease harbored CK (7 abnormalities) by conventional karyotyping and a Tyr234Ser TP53 mutation. He received venetoclax 400 mg daily with 6 doses of monthly rituximab on the phase 1b (M13-365) study37 and achieved a best response of partial remission (PR) at 3 months. After 28 months of therapy, he developed recurrent lymphocytosis and thrombocytopenia, with ∼60% CLL BM infiltration and enlarging nodal disease on computed tomography (CT). Next-generation sequencing identified an additional TP53 mutation (Ser376Lysfs*41) and the BCL2 Gly101Val mutation (variant allele frequency [VAF], 0.4%). We initiated zanubrutinib 160 mg BD on trial and achieved a PR. After 6 months, the BCL2 Gly101Val VAF declined to 0.2%. At 28 months, the patient developed PD, with 3 BTK mutations identified (Leu528Trp, Cys481Ser, and Cys481Tyr). With treatment options limited by impaired patient fitness, we commenced compassionate-access ibrutinib-venetoclax combination and achieved a PR. After 10 months, the patient developed pleural effusions and fluorodeoxyglucose-avid adenopathy on positron emission tomography (PET), with biopsy confirmation of RT (diffuse large B-cell lymphoma [DLBCL]). R-mini-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) was commenced; however, the patient died of respiratory failure on day 21.
How should patients with progressive CLL on venetoclax be managed?
In 2 retrospective series with similar results, BTKi therapy achieved an ORR of 84% to 91% and a median PFS of 32 to 34 months for patients with PD after venetoclax, including patients whose disease harbored BCL2 mutations.33,38 Patients with prior BTKi intolerance who develop venetoclax resistance may achieve durable remissions with BTKi rechallenge, ideally with an alternative agent.38 In contrast, PI3K inhibitors (PI3Kis) after venetoclax are associated with poor outcomes (median PFS, 5 months). At our centers, we have used venetoclax dose escalations up to 600 mg daily in select cases of PD with very limited success.6 In 9 patients with PD on venetoclax monotherapy, additional rituximab deepened responses in 4 cases, with 2 instances of uMRD (including 1 patient with BCL2-mutated disease).
For patients with PD on venetoclax, we advocate BTKi therapy if not previously resistant. For patients previously intolerant to ibrutinib, more selective BTKi's such as acalabrutinib or zanubrutinib may facilitate deliverable therapy with durable remissions. In select cases, additional rituximab and continued venetoclax can deepen responses, whereas dose escalations in this setting are rarely effective. When switching between venetoclax and BTKi's, we continue the first agent until 3 days before the second to avoid potential tumor flare with BTKi cessation.40
How should patients with sequential CLL progression on venetoclax and BTKi's be managed?
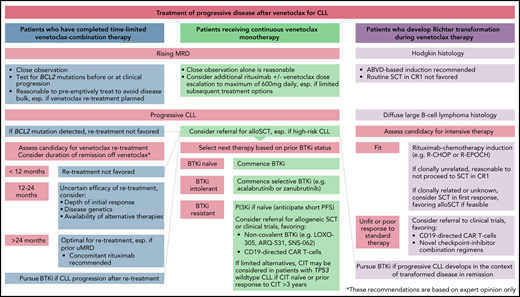
Data informing the treatment of sequentially resistant disease are limited. Outcomes are poor for patients unable to undergo allogeneic SCT (alloSCT), and novel approaches are needed.38 For many clinicians, PI3Kis are the most available next treatment; however, responses are typically short lived.38,41 At our centers, we have used combination ibrutinib-venetoclax therapy for 2 patients with sequentially resistant CLL and have achieved remissions of 10 and 15 months; however, robust data to inform this strategy are lacking.33 One promising agent is pirtobrutinib (LOXO-305), a non-covalent kinase inhibitor with activity against wild-type and Cys481-mutated BTK. In a phase 1/2 study, the ORR in patients with RR CLL was 63%, with 88% of patients still receiving the drug at a median follow-up of 6 months. Response rates were consistent across subgroups previously exposed to BTKi's, venetoclax, or both classes.42 Other noncovalent BTKi's (ARQ-531 and SNS-062) also have activity independent of the Cys481 residue; however, few clinical data are currently available.43,44 A novel BCL2i, BGB-11417, exhibits ∼10-fold greater potency than venetoclax in vitro, and may inhibit Gly101-mutated BCL2 at clinically achievable concentrations.45 A phase 1 trial in mature B-cell malignancies is currently recruiting (www.clinicaltrials.gov, #NCT04277637). Preliminary data are awaited from a phase 1/2 trial in CLL using another BCL2i, APG-2575 (#NCT04494503). Considering the distinct resistance mechanisms between BTKi's and venetoclax,15,21,33,46 there is biological plausibility for more potent BCL2i's against double class-resistant disease and preliminary clinical evidence supporting the non-covalent BTKi pirtobrutinib in this setting42 ; however, longer-term data are needed. Given the paucity of available evidence and the poor results with currently available therapies for these patients, we recommend referral for clinical trials or cellular therapies (case 5) wherever feasible. In the absence of other options, chemoimmunotherapy may be considered for patients with TP53 wild-type CLL who are chemoimmunotherapy naive or have had remissions lasting >3 years after prior chemoimmunotherapy.47 Because of the timing of drug approvals in Australia, most patients treated at our centers have received venetoclax as their first targeted agent and BTKi's subsequently, in contrast to the typical sequence in the United States and other countries. We approach patients with sequentially resistant disease similarly, irrespective of chronology, with modest expectations of PI3Ki treatment and prioritization of clinical trials and cellular therapies where feasible. The reasons for discontinuing prior targeted therapies warrant careful review before deeming patients to have truly double class-resistant disease, as patients who have ceased BTKi's because of intolerance may benefit from more selective agents and those who have received time-limited venetoclax should be considered for retreatment (Figure 2).
Case 2
A 66-year-old man presented with progressive CLL after frontline chlorambucil and subsequent FCR. The disease karyotype was diploid, with no del(17p) detected with fluorescence in situ hybridization (FISH). TP53 mutation testing was not performed. We commenced venetoclax 400 mg daily without rituximab in the phase 1 (M12-175) study8 and achieved a PR by CT at 3 months. PB MRD was monitored every ∼3 months, decreasing to 0.016% at 33 months, followed by a progressive increase. After the patient had received 5 years of venetoclax, we escalated the dose to 600 mg daily, aiming to slow the increasing MRD; however, the increase continued unabated over the ensuing 3 months (PB MRD, 0.44%; BM MRD, 0.78%) without fulfilling International Workshop on Chronic Lymphocytic Leukemia (iwCLL) PD criteria.48 Two BCL2 mutations (Gly101Val, Val156Asp) were detected in a total of 4.4% of CLL cells. Six doses of monthly rituximab were administered, and achieved PB uMRD and 0.014% BM MRD at EOCT. The patient remains on venetoclax at 7-years' follow-up, without PB MRD recrudescence.
How should patients with rising MRD on continuous venetoclax monotherapy be managed?
Although clinical practices vary depending on testing availability, at our centers we generally measure PB MRD every 3 to 6 months in patients receiving venetoclax-based therapy. Using dose escalations up to 600 mg daily, we have attained uMRD in a minority of patients with rising MRD not meeting iwCLL PD criteria.6 As described above, additional rituximab can deepen responses and achieve uMRD in some cases.38 It remains uncertain whether treatment intensification for patients who have MRD persistence or recrudescence offers any advantage over deferring next therapy until clinical PD. Given the evidence implicating alternative BCL2 family proteins in venetoclax resistance,27-30 investigation into the use of BCL-XL or MCL-1 inhibitors to target residual disease is warranted.26,49 For patients with limited subsequent options and rising MRD on continuous venetoclax monotherapy, additional rituximab, with or without dose escalation is reasonable, and can restore deep remissions in some cases.
Case 3
A 71-year-old man treated with FCR 5 years earlier presented with progressive lymphadenopathy and lymphocytosis. The disease karyotype was normal, with no TP53 abnormality detected by sequencing or FISH. He commenced venetoclax 400 mg daily with 6 doses of monthly rituximab within the phase 1b combination study (M13-365)37 and attained a BM MRD− CR after 21 months. Venetoclax was ceased 12 months after BM uMRD attainment. PB MRD recrudescence was detected after 12 months and was serially monitored for 2 years until the development of intraabdominal lymphadenopathy, a ∼50% BM CLL burden and lymphocytosis. BCL2 mutations were not detected. Venetoclax monotherapy was reinstated, achieving a BM MRD+(1.67%) CR at 1 year. The patient remains in remission at last follow-up.
How should patients with progressive CLL after time-limited venetoclax-based therapy be managed?
Fixed-duration venetoclax therapy may remove selection pressure for BCL2-mutated subpopulations, which typically emerge after sustained exposure.21 Furthermore, the action of rituximab and BTKi's against BCL2-mutated CLL suggests that these subclones may be suppressed by upfront combination.33,38 Responses to venetoclax retreatment after time-limited therapy have been recently reported, suggesting that PD in this context frequently retains venetoclax sensitivity.
On longer-term follow-up of the phase 1b venetoclax-rituximab study,37 4 patients underwent retreatment of PD after cessation in deep response. All had ceased drug treatment for >24 months and all responded, with second remissions ranging from 19 to 40+ months (2 remissions ongoing; Shuo Ma, John F. Seymour, Danielle M. Brander, Thomas J. Kipps, Michael Y. Choi, Mary Ann Anderson, Kathryn Humphrey, Abdullah Al Masud, Ruby Nandam, Ahmed Hamed Salem, Brenda Chyla, Jennifer Arzt, Amanda Jacobson, Su Young Kim, and Andrew W. Roberts, manuscript submitted May 2021). In a retrospective analysis of patients with mostly BTKi-exposed CLL and a median time of initial venetoclax therapy of 9 months, the ORR to retreatment was 72% (CRR, 22%) among 18 response-evaluable subjects. Most patients (80%) were retreated with venetoclax, with or without anti-CD20 antibody, and the estimated 1-year PFS was 69%.50 In an update from the MURANO study, among 18 response-evaluable patients with a median time of venetoclax of 24 months, the ORR to retreatment was 72%, with 50% of patients remaining on therapy after a median of 11 months. In the same report, all 14 patients who received BTKi's for PD after venetoclax-rituximab responded, with 71% continuing therapy at a median of 22 months.51 These data demonstrate that venetoclax-based retreatment achieves frequent responses, although longer-term follow-up is necessary to evaluate remission durability.
Where available, we recommend assessment for BCL2 mutations in patients with increasing MRD or PD after time-limited venetoclax. If BCL2 mutations are not identified, we consider venetoclax retreatment, preferably with concomitant anti-CD20 antibody. Standard ramp-up and tumor lysis risk management strategies apply.52 Treatment should be initiated for iwCLL indications, but may be introduced earlier if necessary to avoid the development of disease bulk and allow for dose ramp-up without urgency.48 Given that the median time to clinical PD after MRD recrudescence was >2 years in the MURANO trial, we do not recommend retreatment of MRD conversion alone.11 Prolonged time off initial venetoclax-based therapy is anticipated to predict the greatest benefit from retreatment, however informative data are currently lacking. Awaiting further evidence, we do not favor retreatment of patients who have ceased venetoclax for <12 months before developing PD, unless no other feasible therapeutic options exist. We consider patients with treatment-free remissions lasting >24 months, especially if uMRD was achieved with first exposure, to be ideal candidates for retreatment. In cases of intermediate time of therapy (12-24 months), the initial response depth, genetic risk profile and availability of alternative therapies should be considered.
It is unknown whether patients who attain uMRD after retreatment can again cease therapy and maintain disease control. Patients with PD after retreatment should receive BTKi's if previously naive, or be considered for cellular therapy or trials. Proceeding to BTKi's without attempting retreatment is reasonable and associated with high ORRs; however, given the limited therapeutic options for double class-resistant CLL, we currently favor retreatment to maximize the clinical benefit from venetoclax-based therapy.
Case 4
A 62-year-old woman presented with bulky CLL after 2 prior fludarabine-based regimens (relapsing <6 months after second-line therapy) and R-CHEP (rituximab, cyclophosphamide, doxorubicin, etoposide, and prednisolone). Her disease harbored del(17p) by FISH, without CK. She received venetoclax 200 mg daily (assigned cohort dose) in the phase 1 (M12-175) trial8 attaining a PR. At 16 months, biopsy of an enlarged tonsillar node detected on surveillance CT confirmed DLBCL-RT, with stage IV disease on PET. IGHV analysis was not performed (clonal relationship to CLL unknown). No suitable allogeneic donor was available. We delivered 2 cycles of R-ICE (rituximab, ifosfamide, carboplatin, and etoposide) followed by a high-dose CBV (cyclophosphamide, carmustine, and etoposide) preparative regimen and autologous SCT (autoSCT), and achieved a complete metabolic response. Nineteen months after autograft the patient developed progressive CLL. Repeat cytogenetics identified new CK (eleven abnormalities) and persistent del(17p). We initiated ibrutinib 420 mg daily achieving a PR. After 3 years of receiving ibrutinib, she presented with rapidly progressive DLBCL. Venetoclax was added to ibrutinib for 1 month with a mixed response. Salvage R-CHOP was complicated by sepsis and the patient died.
How should patients with RT on venetoclax be managed?
RT typically occurs early in venetoclax therapy (<24 months), particularly among patients with heavily pretreated disease refractory to fludarabine or bearing CK.34 We perform PET imaging before venetoclax in these high-risk subsets and biopsy sites with a maximum standard uptake value >5 whenever feasible, although the specificity of this threshold is low.53,54 As in other contexts, the prognosis of DLBCL-RT emergent during venetoclax therapy is poor, although durable responses can be achieved for a minority of patients.34 In the ∼20% of DLBCL unrelated to the underlying CLL, it is reasonable to treat as de novo disease, although data in the context of targeted agents are lacking.55 Where clonal relatedness is unknown, the ORR to R-CHOP or R-EPOCH (rituximab, etoposide, prednisolone, vincristine, cyclophosphamide, and doxorubicin) is 40% to 60%, but remissions are typically short lived without SCT.56,57 Where feasible, we favor reduced intensity alloSCT because of its proven curative potential and the inevitable CLL relapse after autoSCT.58 In selected patients who were unable to proceed to alloSCT, we have achieved durable survival using salvage chemotherapy with or without autoSCT followed by BTKi's for subsequent CLL relapse.33 Given the limited therapeutic options, we recommend referral for clinical trials where available. Preliminary results for CD19-directed chimeric antigen receptor (CAR) T-cell therapy for patients with RT after targeted agents are promising, but mature data from larger cohorts are required.59,60 BTKi or immune-checkpoint inhibitor monotherapy have achieved modest ORRs in small cohorts, but CRs are infrequent and survival is poor.61-62 In a phase 2 trial of 23 patients with RT, the combination of nivolumab and ibrutinib achieved an ORR of 43% (CRR: 35%), although the median remission duration was short (9.3 months).63 For the rarer Hodgkin-type RT, outcomes are more favorable. In a retrospective study, 62 patients with Hodgkin-type RT received ABVD (doxorubicin, bleomycin, vinblastine, decarbazine)−based therapy, largely without transplantation and had a median OS of ∼13 years.64 We have observed similarly favorable results in patients with Hodgkin-type RT emergent on venetoclax.34 For these patients, we recommend ABVD-based therapy without SCT in CR1.
Case 5
A 47-year-old man previously treated with FCR, R-CHOP, and navitoclax26 was enrolled in the venetoclax phase 1 (M12-175) study.8 Pretreatment genetic analyses identified del(17p) and CK (5 abnormalities). After achieving a PR, the patient developed progressive lymphadenopathy on surveillance CT at 16 months, with histological confirmation of DLBCL-RT. No suitable allogeneic donor was available at this time. Four cycles of R-ICE followed by a busulfan/melphalan autoSCT achieved a CR, but progressive CLL developed after 1 year. Repeat cytogenetics detected 6 new abnormalities that had not been observed. We commenced zanubrutinib on trial, achieving a MRD+ CR at 2 years. The patient developed progressive CLL after 30 months of zanubrutinib, and idelalisib-rituximab was initiated, with PR achieved after 5 months. Given the anticipated short-lived response, the patient underwent a fludarabine/melphalan/thymoglobulin, single-antigen mismatched unrelated donor alloSCT, with 100% donor chimerism at day 30. After 1 year, BM MRD was undetectable, but surveillance PET identified a fluorodeoxyglucose-avid para-aortic node, histologically confirmed to be DLBCL. Involved field radiotherapy achieved a CR, and we commenced interferon to enhance the graft-versus-leukemia effect. At 18 months after alloSCT, the patient developed a mandibular lesion confirmed to be accelerated CLL. The investigational use of nivolumab was unsuccessful; however, radiotherapy achieved a near CR at last response assessment. He remains alive 7 years after venetoclax-resistant RT diagnosis without graft-versus-host disease (GVHD).
What is the role of cellular therapies in the targeted agent era?
For patients with RR CLL, the median PFS after venetoclax-rituximab or ibrutinib as first targeted agent is 4 to 5 years11,25 and 2 to 3 years after the second.15,33,38 Despite these advances, young patients or those with TP53-aberrant disease are likely to develop double class resistance with limited therapeutic options. Recent data have confirmed that reduced-intensity alloSCT remains effective after targeted agents. In retrospective study of 65 patients exposed to targeted agents, the 2-year PFS and OS after alloSCT were 63% and 81%.65 A similar study of 30 patients with targeted agent–exposed, high-risk CLL reported a 3-year PFS and OS of 72% and 87% after alloSCT.66 Both groups reported modest nonrelapse mortality rates (7% to 13%), with moderate acute GVHD (grades 3-4, 13% to 24%) and chronic GVHD (27% to 57%). In both studies, higher hematopoietic cell transplantation comorbidity index was associated with inferior survival, whereas adverse disease genetics, response status, and number of prior targeted agents were not.65,66 We consider alloSCT for eligible patients who (1) have CLL resistant to the first targeted agent (BTKi or venetoclax), (2) are in good response with the first targeted agent for high-risk disease (TP53 aberrant, CK, fludarabine-refractory, or heavily pretreated), and (3) are in remission after induction for DLBCL-RT, especially if clonal relatedness is proven.
CAR T cells are a promising therapeutic approach in CLL. In phase 1/2 studies of CD19-directed CAR T-cell therapy for patients with genetically adverse, ibrutinib-exposed disease, BM MRD negativity by IGH sequencing (sensitivity, 10−6) was achieved in 58% to 76% of evaluable patients, although current follow-up is short.67,68 Phase 1/2 studies combining ibrutinib with CD19-directed CAR T cells in small cohorts of targeted agent–resistant CLL have achieved BM uMRD by IGH sequencing in >60% of evaluable patients, including subsets with dual BTKi-venetoclax resistant CLL.69-70 Compared with historical controls, concomitant ibrutinib may be associated with lower rates of cytokine release syndrome (CRS), despite comparable CAR T-cell expansion, potentially due to interleukin-2–inducible T-cell kinase inhibition and downregulation of CRS-associated cytokines.69 Fatal cardiac arrhythmias during CRS and neurotoxicity have been reported in 2 patients in these early trials, with potential contribution from the arrhythmogenic properties of ibrutinib, raising an important safety signal to monitor in future studies.67,69,71 Overall, CAR T-cell therapy can achieve deep remissions in high-risk, targeted agent–resistant disease; however, the current evidence is derived from small, selected cohorts, and the generalizability of their results is uncertain. Although prolonged remissions have been reported,72 longer-term follow-up of larger cohorts is required to meaningfully evaluate the durability of initial deep responses. For patients unsuitable for alloSCT because of fitness, donor availability, or preference, referral for CAR T-cell therapy on trial is worthwhile.
Conclusions
The capacity to induce deep responses positions venetoclax as a central component of combination targeted agent therapy for CLL. Unfortunately, most patients with RR disease ultimately develop relapse, typified by oligoclonal PD with multiple distinct resistance mechanisms. Our approach to the treatment of PD after venetoclax is summarized in Figure 2. With substantial progress in the targeted agent era, many outstanding questions emerge. The survival mechanisms within the early clones which propagate venetoclax-resistant disease are unknown, as are therapies to undermine them. The resistance mechanisms identified after continuous targeted agent monotherapy are likely to differ from those for patients receiving upfront, time-limited, combination regimens, requiring significant further investigation. Worldwide, clinicians will increasingly encounter patients with dual BTKi-venetoclax−resistant disease, for which the resistance biology and optimal treatment remain areas of much needed further research.
Acknowledgments
The authors thank Mary Ann Anderson, Amit Khot, and Henry Januszewicz who also provided care to the patients in these vignettes; Piers Blombery, who characterized the BCL2 and TP53 mutations in several of the above cases; and the patients, their family members, and the members of the staff who participated in early clinical trials of venetoclax.
Authorship
Contribution: T.E.L., C.S.T., and J.F.S. contributed equally to the writing of the manuscript.
Conflict-of-interest disclosure: T.E.L. is an employee of the Walter and Eliza Hall Institute of Medical Research, which receives milestone and royalty payments related to venetoclax, is a recipient of a share in royalty payments paid to the Walter and Eliza Hall Institute of Medical Research, and has received honoraria from AbbVie. C.S.T. has received honoraria and research funding from AbbVie and Janssen and honoraria from BeiGene. J.F.S. has received research funding from AbbVie, Genentech, Celgene, and Janssen and is an advisory board member for and has received honoraria from AbbVie, Acerta, Celgene, Genentech, Janssen, Roche, Sunesis, and Takeda.
Correspondence: John F. Seymour, Department of Clinical Haematology, The Royal Melbourne Hospital and Peter MacCallum Cancer Centre, Melbourne, VIC 3000, Australia; e-mail: john.seymour@petermac.org.
