

Key Points
SARS-CoV-2 spike protein pseudovirus infection enhances TF procoagulant activity and releases TF+ EVs via activation of ASMase.
FDA-approved tricyclic antidepressants that act as functional inhibitors of ASMase attenuate the pseudovirus-induced enhanced TF activity.

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection is associated with the hypercoagulable state. Tissue factor (TF) is the primary cellular initiator of coagulation. Most of the TF expressed on cell surfaces remains cryptic. Sphingomyelin (SM) is responsible for maintaining TF in the encrypted state, and hydrolysis of SM by acid sphingomyelinase (ASMase) increases TF activity. ASMase was shown to play a role in virus infection biology. In the present study, we investigated the role of ASMase in SARS-CoV-2 infection-induced TF procoagulant activity. Infection of human monocyte–derived macrophages (MDMs) with SARS-CoV-2 spike protein pseudovirus (SARS-CoV-2–SP-PV) markedly increased TF procoagulant activity at the cell surface and released TF+ extracellular vesicles. The pseudovirus infection did not increase either TF protein expression or phosphatidylserine externalization. SARS-CoV-2–SP-PV infection induced the translocation of ASMase to the outer leaflet of the plasma membrane, which led to the hydrolysis of SM in the membrane. Pharmacologic inhibitors or genetic silencing of ASMase attenuated SARS-CoV-2–SP-PV–induced increased TF activity. Inhibition of the SARS-CoV-2 receptor, angiotensin-converting enzyme-2, attenuated SARS-CoV-2–SP-PV–induced increased TF activity. Overall, our data suggest that SARS-CoV-2 infection activates the coagulation by decrypting TF through activation of ASMase. Our data suggest that the US Food and Drug Administration–approved functional inhibitors of ASMase may help treat hypercoagulability in patients with COVID-19.
Introduction
Although respiratory dysfunction is the primary feature of the COVID-19, many studies showed that a hypercoagulability state is frequently present in patients with COVID-19.1,2 Thromboembolic complications play a crucial role in the pathogenesis and mortality of COVID-19.3,4 Aberrant expression of tissue factor (TF) is associated with many forms of thrombosis.5 Recent reviews suggest that TF may play a central role in thrombosis in patients with COVID-19.6,7 Increased expression of TF was found in platelet-monocyte aggregates from critically ill patients with COVID-19.8 Skendros et al9 showed the expression of TF in neutrophils of patients with severe COVID-19. However, in the above studies, TF expression analyses were limited to microscopy, flow cytometry, or mRNA levels. Recently, Rosell et al10 reported that levels of extracellular vesicle (EV) TF activity were significantly higher in patients with COVID-19 than control subjects, and the levels of EV TF activity were associated with disease severity and mortality. Interestingly, in contrast to the above studies, Mast et al11 reported no increase in TF mRNA transcript in bronchoalveolar lavage fluids of patients with COVID-19.
TF expressed on cells, either constitutively or induced, possess very low or no procoagulant activity.12 TF has to undergo posttranslational modifications, commonly referred to as decryption or activation, to exhibit optimal TF procoagulant activity.12 Our recent studies showed that high sphingomyelin (SM) content in the outer leaflet of plasma membrane maintains TF in an encrypted state, and the hydrolysis of SM induced by pathophysiologic stimuli leads to TF activation.13,14 The breakdown of SM in the plasma membrane also leads to the generation of EVs.13,14 Many stimuli, including viral infections, can release sphingomyelinases, such as acid sphingomyelinase (ASMase), that can breakdown SM in the plasma membrane.15 Recent studies suggest that ASMase plays a role in the infectivity of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).16 Here, we investigated the role of ASMase in SARS-CoV-2 infection–induced TF-mediated coagulation.
Study design
Generation and titration of SARS-CoV-2 spike protein enveloped pseudovirus (SARS-CoV-2–SP-PV)
Two hundred ninety-three T cells (1 × 107 cells) were transfected with 20 μg vector plasmid pLVX-IRES-ZsGreen (Takara Bio), 16 μg packaging plasmid psPAX2 (Addgene), and 10 μg envelope plasmid pCAGGS–SARS-CoV-2 spike (obtained from BEI Resources) using polyethyleneimine (Polysciences, Warrington, PA). After 30 minutes of transfection, Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum and antibiotics was added to the cells, and the cells were cultured at 37°C under 5% CO2. After 48 to 72 hours, the supernatants were collected, filtered, and centrifuged at 25 000g for 2.5 hours at 4°C to sediment the pseudovirus particles. The pseudovirus pellet was resuspended in phosphate-buffered saline, and the viral titers were determined by infecting a human lung cell line, H441, with serially diluted pseudovirus preparations.
Generation of human monocyte–derived macrophages and isolation of extracellular vesicles
The Institutional Review Board at the University of Texas Health Science Center at Tyler approved the protocol to obtain blood from healthy volunteers. Human peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation, and monocytes were differentiated into macrophages, as described earlier.13,14 EVs from cell supernatants were isolated by centrifugation at 21 000g for 1 hour and quantified by nanoparticle tracking analysis as described recently.13,17
Measurement of TF activity
TF activity on cell surfaces and EVs was measured as its ability to support the activation of factor X (FX) on the addition of FVIIa and FX.13,14
Immunofluorescence microscopy and image acquisition
Immunofluorescence labeling of TF, ASMase, and SM in monocyte–derived macrophages (MDMs), immunofluorescence confocal microscopy, and image acquisition were performed essentially as described in our earlier publications.13,14
Inhibition of ASMase or ACE-2 by siRNA
Macrophages were transfected with a control transfection reagent (mock transfection), scrambled oligonucleotide (scRNA), or small interfering RNA (siRNA) specific for ASMase (100 nM) or ACE-2 (100 nM) using X-tremeGene transfection reagent (Sigma-Aldrich, St Louis, MO).
Results and discussion
SARS-CoV-2–SP-PV infection leads to increased cell surface TF activity and release of TF+ EVs
Infection of MDMs with SARS-CoV-2–SP-PV (0.6 multiplicity of infection) led to a marked increase in cell surface TF activity in a time-dependent manner. The increased cell surface TF activity was evident as early as 5 to 15 minutes after the infection but markedly higher at 6 hours after infection (Figure 1A). SARS-CoV-2–SP-PV infection also increased TF activity associated with EVs, but the increase plateaued after 30 minutes (Figure 1A). EVs isolated from the supernatant medium from 0. 5 to 6 hours after infection showed three- to fivefold higher TF activity compared with EVs isolated from MDMs treated with a control vehicle or a control pseudovirus. In additional studies, we quantified the number of EVs released from MDMs after infection with SARS-CoV-2–SP-PV by nanoparticle tracking analysis. SARS-CoV-2–SP-PV infection increased the number of EVs released from MDMs in a time-dependent manner up to 30 minutes, and after that, no further increase in EVs number was observed (supplemental Figure 1, available on the Blood Web site). The mean diameter of EVs was 148.3 ± 30.1 nm. The fold increase in the number of EVs was very similar to the fold increase observed with EV-associated TF activity in SARS-CoV-2–SP-PV–infected cells (Figure 1A). No significant differences in EVs number between 30 minutes and 6 hours may reflect the balance between EVs release from MDMs and the uptake of EVs by the same MDMs. Evaluation of total TF protein expression levels by immunoblot analysis (Figure 1B) or cell surface TF expression by flow cytometry (Figure 1C) or immunofluorescence confocal microscopy (Figure 1D) showed that SARS-CoV-2–SP-PV infection did not increase TF protein expression levels. Treatment of intact cells or EVs with TF antibodies fully attenuated the increased FXa generation associated with SARS-CoV-2–SP-PV infection (Figure 1E). Externalization of phosphatidylserine (PS) following the injury is believed to be primarily responsible for increased TF activity.12,18 However, annexin V pretreatment, which blocks PS-dependent increased TF activity, failed to attenuate the pseudovirus-induced TF activity (Figure 1F). Consistent with this observation, we found no detectable increase in the binding of AF488–annexin V to the pseudovirus-infected macrophages (Figure 1G). Taken together, these results suggest that SARS-CoV-2 infection is likely to activate TF-mediated coagulation by decrypting TF and releasing TF+ EVs into circulation, without inducing de novo synthesis of TF or the externalization of PS. Consistent with these data, we found that SARS-CoV-2–SP-PV infection did not increase the procoagulant activity of naïve PBMCs that do not constitutively express TF protein but enhanced the procoagulant activity of lipopolysaccharide-stimulated PBMCs that express TF protein (supplemental Figure 2).
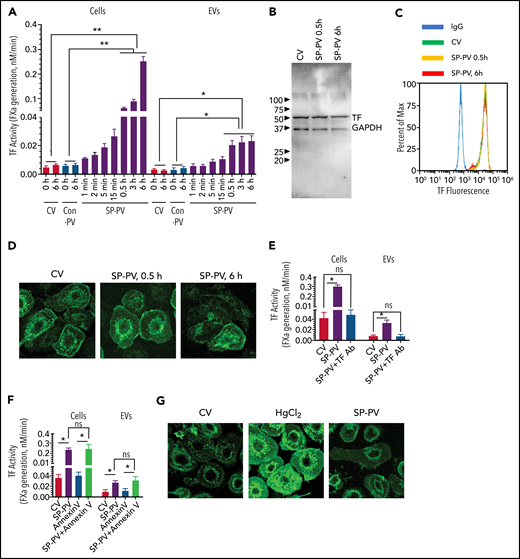
SARS-CoV-2–SP-PV infection increases cell surface TF activity and releases TF+ vesicles in human MDMs. (A) MDMs were treated with control vehicle (CV) or infected with SARS-CoV-2–SP-PV (SP-PV) or vesicular stomatitis virus G protein envelope pseudotyped lentivirus, which was generated and isolated in the same fashion as of SARS-CoV-2–SP-PV, as a control pseudovirus (Con-PV). At the indicated time, the cell supernatants were removed, and the EVs released into the conditioned medium were isolated by centrifugation at 21 000g for 1 hour. TF activity associated with the cell surface or EVs was determined by adding FVIIa (10 nM) and the substrate factor X (175 nM) and measuring the rate of factor Xa generation in a chromogenic assay. (B-D) Analysis of total and cell surface TF protein expression in MDMs treated with CV or infected with SARS-CoV-2–SP-PV by immunoblot analysis (B), flow cytometry (C), or immunofluorescence confocal microscopy (D). (E-F) Intact MDMs infected with SARS-CoV-2–SP-PV (for 6 hours) or EVs released from the infected MDMs were incubated with polyclonal rabbit anti-human TF antibody (10 µg/mL) for 1 hour (E) or annexin V (400 nM) for 30 minutes (F) before measuring cell surface– or EV-associated TF activity as described in panel A. (G) SARS-CoV-2–SP-PV infection does not induce the externalization of phosphatidylserine. MDMs were treated with a control vehicle or infected with the pseudovirus for 6 hours and then stained with AF488–annexin V and subjected to fluorescence microscopy. As a positive control, MDMs were treated with HgCl2 (5 µg/mL) for 5 minutes to externalize phosphatidylserine. All experiments were repeated at least 3 times, and data were expressed as mean ± standard error of the mean (SEM). Statistical significance between the 2 groups was calculated using the Mann-Whitney test and among the groups by 1-way analysis of variance followed by Tukey’s post hoc analysis. *P < .05; **P < .01; ns, not statistically significant (compared with values obtained with control vehicle treatment).
SARS-CoV-2–SP-PV infection increases cell surface TF activity and releases TF+ vesicles in human MDMs. (A) MDMs were treated with control vehicle (CV) or infected with SARS-CoV-2–SP-PV (SP-PV) or vesicular stomatitis virus G protein envelope pseudotyped lentivirus, which was generated and isolated in the same fashion as of SARS-CoV-2–SP-PV, as a control pseudovirus (Con-PV). At the indicated time, the cell supernatants were removed, and the EVs released into the conditioned medium were isolated by centrifugation at 21 000g for 1 hour. TF activity associated with the cell surface or EVs was determined by adding FVIIa (10 nM) and the substrate factor X (175 nM) and measuring the rate of factor Xa generation in a chromogenic assay. (B-D) Analysis of total and cell surface TF protein expression in MDMs treated with CV or infected with SARS-CoV-2–SP-PV by immunoblot analysis (B), flow cytometry (C), or immunofluorescence confocal microscopy (D). (E-F) Intact MDMs infected with SARS-CoV-2–SP-PV (for 6 hours) or EVs released from the infected MDMs were incubated with polyclonal rabbit anti-human TF antibody (10 µg/mL) for 1 hour (E) or annexin V (400 nM) for 30 minutes (F) before measuring cell surface– or EV-associated TF activity as described in panel A. (G) SARS-CoV-2–SP-PV infection does not induce the externalization of phosphatidylserine. MDMs were treated with a control vehicle or infected with the pseudovirus for 6 hours and then stained with AF488–annexin V and subjected to fluorescence microscopy. As a positive control, MDMs were treated with HgCl2 (5 µg/mL) for 5 minutes to externalize phosphatidylserine. All experiments were repeated at least 3 times, and data were expressed as mean ± standard error of the mean (SEM). Statistical significance between the 2 groups was calculated using the Mann-Whitney test and among the groups by 1-way analysis of variance followed by Tukey’s post hoc analysis. *P < .05; **P < .01; ns, not statistically significant (compared with values obtained with control vehicle treatment).
Role of ASMase in SARS-CoV-2–SP-PV-induced TF activation
Many stress indicators, including viral infections, induce translocation of ASMase from intracellular compartments onto the outer leaflet of the plasma membrane.19 ASMase expression in MDMs treated with a control vehicle was limited primarily to the intracellular compartment (Figure 2A, top images). SARS-CoV-2–SP-PV infection mobilized ASMase from intracellular compartments to the outer leaflet of the plasma membrane (Figure 2A, white arrowheads) without altering total ASMase protein levels (supplemental Figure 3). Cell surface SM staining with lysenin showed a marked decrease in SM in SARS-CoV-2–SP-PV–infected cells compared with cells treated with a control vehicle (Figure 2A, bottom images). These data indicate that SARS-CoV-2 infection induces the activation ASMase and reduces SM content in the plasma membrane. To determine the role of ASMase in enhancing TF activity in MDMs infected with SARS-CoV-2–SP-PV, MDMs were treated with functional inhibitors of ASMase or ASMase expression was silenced by siRNA. Pretreatment of MDMs with ASMase functional inhibitors, desipramine and imipramine, blocked the pseudovirus-induced translocation of ASMase to the plasma membrane (supplemental Figure 4). Treatment of MDMs with ASMase functional inhibitors or ASMase silencing by siRNA curtailed the pseudovirus-induced increased TF activity on the cell surface and the release of TF+ EVs (Figure 2B-C). Consistent with our earlier published data,13,14 inhibition of ASMase did not affect TF protein expression levels (supplemental Figure 5A).
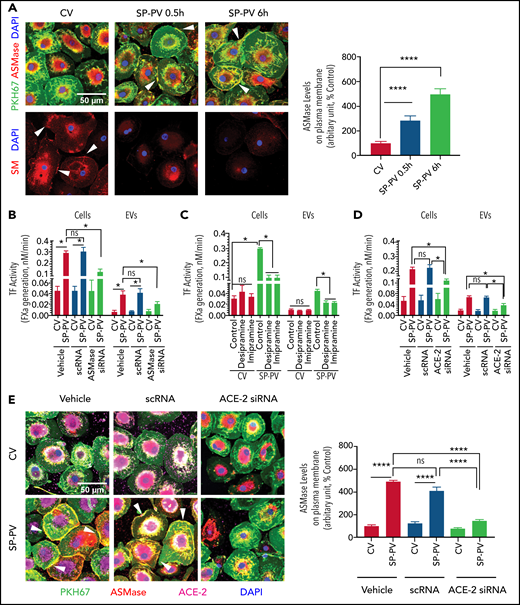
SARS-CoV-2–SP-PV–induced increased TF activity is dependent on ASMase and ACE2. (A) SARS-CoV-2–SP-PV infection induces the translocation of ASMase to the cell surface and reduces SM content in the plasma membrane. MDMs were infected with a control vehicle (CV) or infected with SARS-CoV-2–SP-PV (SP-PV) for 0.5 or 6 hours. Top of image: plasma membrane was stained with PKH67 fluorescent dye (green). For immunostaining ASMase, fixed and permeabilized cells were incubated with rabbit anti-human ASMase antibody (2 µg/mL) at 4°C overnight. After removing the primary antibodies and washing the cells, the cells were incubated with AF546-conjugated donkey anti-rabbit IgG (2 µg/mL) for 90 minutes (red). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; blue). The yellow color in the plasma membrane (white arrow marks) represents the colocalization of ASMase staining with PKH67 fluorescence in the plasma membrane, indicating the translocation of ASMase to the outer leaflet of the plasma membrane. The fluorescence signaling intensity of ASMase in the plasma membrane was quantified by choosing 30 random areas in the plasma membrane and using ImageJ software (National Institutes of Health). The quantified data were shown in a bar graph. Bottom of image: to label plasma membrane SM, fixed and nonpermeabilized cells were incubated with an SM-binding protein, lysenin (1 µg/mL), for 2 hours at room temperature, and then cells were incubated with rabbit anti-human lysenin antibody (1:200) overnight, followed by secondary antibody, AF546-conjugated donkey anti-rabbit IgG for 90 minutes (red). Nuclei were stained with DAPI (blue). The immunofluorescence staining of SM (red) was analyzed by confocal microscopy. White arrowheads point out SM in the outer plasma membrane in control (noninfected) cells. (B) ASMase silencing attenuates the SARS-CoV-2–SP-PV–induced increased TF activity. MDMs were transfected with transfection reagent (vehicle), scrambled oligonucleotide (scRNA), or siRNA specific for ASMase (100 nM) using X-tremeGene transfection reagent. After 48 hours, transfected MDMs were infected with the pseudovirus for 6 hours, and EVs released into the supernatant medium were isolated. Cell surface TF activity and TF activity associated with EVs were measured as described in panel C. (C) ASMase functional inhibitors attenuate SARS-CoV-2–SP-PV–induced increased TF activity in MDMs. MDMs were pretreated with an ASMase inhibitor, desipramine (1 μM) or imipramine (1 μM), for 1 hour and then infected with the pseudovirus for 6 hours. EVs released into the supernatant medium were isolated. TF activity on the cell surface and associated with EVs was determined by adding FVIIa (10 nM) and the substrate factor X (175 nM) and measuring the rate of factor Xa generation. (D) ACE-2 silencing attenuates SARS-CoV-2–SP-PV–induced increased TF activity. MDMs were transfected with transfection reagent (vehicle), scrambled oligonucleotide (scRNA), or siRNA specific for ACE-2 (100 nM). After 48 hours, the transfected MDMs were infected with the pseudovirus for 6 hours, and EVs released into the supernatant medium were isolated. TF activity on cell surfaces or EVs was measured as described for panel C. (E) ACE-2 silencing blocks SARS-CoV-2–SP-PV–induced ASMase translocation to the outer leaflet of the plasma membrane. MDMs transfected with transfection reagent (vehicle), scrambled oligonucleotide (scRNA), or siRNA specific for ACE-2 (100 nM) were infected with the pseudovirus for 6 hours. After that, cells were fixed, permeabilized, and stained with rabbit anti-human ASMase antibody as described in panel A (red). Cells were also immunostained for ACE-2 (magenta). Nuclei were stained with DAPI (blue) and plasma membrane was stained with PKH67 fluorescent dye (green). White arrowheads point out ASMase translocated to the plasma membrane in the vehicle-treated or scRNA-transfected cells that were infected with the pseudovirus. The fluorescence signaling intensity of ASMase staining in the plasma membrane was quantified using Image J software. These data were shown in a bar graph. Statistical significance between the 2 groups was calculated using the Mann-Whitney test. *P < .05; ****P < .0001; ns, not statistically significant.
SARS-CoV-2–SP-PV–induced increased TF activity is dependent on ASMase and ACE2. (A) SARS-CoV-2–SP-PV infection induces the translocation of ASMase to the cell surface and reduces SM content in the plasma membrane. MDMs were infected with a control vehicle (CV) or infected with SARS-CoV-2–SP-PV (SP-PV) for 0.5 or 6 hours. Top of image: plasma membrane was stained with PKH67 fluorescent dye (green). For immunostaining ASMase, fixed and permeabilized cells were incubated with rabbit anti-human ASMase antibody (2 µg/mL) at 4°C overnight. After removing the primary antibodies and washing the cells, the cells were incubated with AF546-conjugated donkey anti-rabbit IgG (2 µg/mL) for 90 minutes (red). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; blue). The yellow color in the plasma membrane (white arrow marks) represents the colocalization of ASMase staining with PKH67 fluorescence in the plasma membrane, indicating the translocation of ASMase to the outer leaflet of the plasma membrane. The fluorescence signaling intensity of ASMase in the plasma membrane was quantified by choosing 30 random areas in the plasma membrane and using ImageJ software (National Institutes of Health). The quantified data were shown in a bar graph. Bottom of image: to label plasma membrane SM, fixed and nonpermeabilized cells were incubated with an SM-binding protein, lysenin (1 µg/mL), for 2 hours at room temperature, and then cells were incubated with rabbit anti-human lysenin antibody (1:200) overnight, followed by secondary antibody, AF546-conjugated donkey anti-rabbit IgG for 90 minutes (red). Nuclei were stained with DAPI (blue). The immunofluorescence staining of SM (red) was analyzed by confocal microscopy. White arrowheads point out SM in the outer plasma membrane in control (noninfected) cells. (B) ASMase silencing attenuates the SARS-CoV-2–SP-PV–induced increased TF activity. MDMs were transfected with transfection reagent (vehicle), scrambled oligonucleotide (scRNA), or siRNA specific for ASMase (100 nM) using X-tremeGene transfection reagent. After 48 hours, transfected MDMs were infected with the pseudovirus for 6 hours, and EVs released into the supernatant medium were isolated. Cell surface TF activity and TF activity associated with EVs were measured as described in panel C. (C) ASMase functional inhibitors attenuate SARS-CoV-2–SP-PV–induced increased TF activity in MDMs. MDMs were pretreated with an ASMase inhibitor, desipramine (1 μM) or imipramine (1 μM), for 1 hour and then infected with the pseudovirus for 6 hours. EVs released into the supernatant medium were isolated. TF activity on the cell surface and associated with EVs was determined by adding FVIIa (10 nM) and the substrate factor X (175 nM) and measuring the rate of factor Xa generation. (D) ACE-2 silencing attenuates SARS-CoV-2–SP-PV–induced increased TF activity. MDMs were transfected with transfection reagent (vehicle), scrambled oligonucleotide (scRNA), or siRNA specific for ACE-2 (100 nM). After 48 hours, the transfected MDMs were infected with the pseudovirus for 6 hours, and EVs released into the supernatant medium were isolated. TF activity on cell surfaces or EVs was measured as described for panel C. (E) ACE-2 silencing blocks SARS-CoV-2–SP-PV–induced ASMase translocation to the outer leaflet of the plasma membrane. MDMs transfected with transfection reagent (vehicle), scrambled oligonucleotide (scRNA), or siRNA specific for ACE-2 (100 nM) were infected with the pseudovirus for 6 hours. After that, cells were fixed, permeabilized, and stained with rabbit anti-human ASMase antibody as described in panel A (red). Cells were also immunostained for ACE-2 (magenta). Nuclei were stained with DAPI (blue) and plasma membrane was stained with PKH67 fluorescent dye (green). White arrowheads point out ASMase translocated to the plasma membrane in the vehicle-treated or scRNA-transfected cells that were infected with the pseudovirus. The fluorescence signaling intensity of ASMase staining in the plasma membrane was quantified using Image J software. These data were shown in a bar graph. Statistical significance between the 2 groups was calculated using the Mann-Whitney test. *P < .05; ****P < .0001; ns, not statistically significant.
Spike protein binds angiotensin-converting enzyme-2 (ACE-2) on host cells, which allows viral entry into cells.20,21 To determine whether SARS-CoV-2–SP-PV-induced ASMase activation, TF decryption, and the release of EVs require spike protein binding to ACE-2, we silenced ACE-2 expression in MDMs (supplemental Figure 5B). ACE-2 silencing did not affect TF protein expression (supplemental Figure 5C-D) but attenuated the pseudovirus-induced increased TF activity at the cell surface as well as the release of TF+ EVs (Figure 2D). Analysis of ASMase expression by immunofluorescence confocal microscopy showed that ACE-2 silencing attenuated the pseudovirus-induced ASMase translocation to the plasma membrane (Figure 2E). These results suggest that SARS-CoV-2–SP-PV enhances TF activity via its interaction with ACE-2 and the subsequent activation of ASMase.
Earlier studies with rhinovirus22,23 and measles virus24 indicate that the hydrolysis of SM and ceramide generation plays a key role in viral entry. Recent studies show that infection of epithelial cells with SARS-CoV-2 or the pseudovirus containing spike protein activates ASMase within 20 to 30 minutes, and pharmacologic inhibition of ASMase prevents the virus entry into the cells.16,25 In earlier studies, we showed that activation of ASMase plays a critical role in TF decryption and release of EVs.13,14 Therefore, it is likely that SARs-CoV-2 infection activates TF-mediated coagulation through TF decryption and the release of TF+ EVs through activation of ASMase and the subsequent breakdown of SM. It is not feasible in our studies to differentiate whether the observed effect of ASMase inhibitors in attenuating SARS-CoV-2–SP-PV–induced TF decryption comes from the inhibition of the pseudovirus-induced ASMase activation or blockade of the pseudovirus entry into the cells. Because ASMase inhibitors also blocked purified spike protein-induced TF decryption (data not shown), ASMase inhibitors would likely block SARS-CoV-2–induced TF activation independent of their effect on the viral entry. In summary, our data raise the possibility that US Food and Drug Administration–approved functional inhibitors of ASMase may help in preventing or treating COVID-19–associated coagulopathy.
Acknowledgments
This work was supported by National Institutes of Health grants R01HL124055 from the National Heart, Lung, and Blood Institute (L.V.M.R.) and UG3AI150550 from the National Institute of Allergy and Infectious Diseases (G.Y.), and a postdoctoral fellowship award (19POST34380330) from the American Heart Association (J.W.).
Authorship
Contribution: J.W. performed experiments, summarized the data, and wrote the initial draft of the manuscript; G.Y. provided SARS-CoV-2 pseudovirus; U.R.P. contributed to designing experiments, reviewing data, and editing the manuscript; L.V.M.R. conceived and designed the study, analyzed the data, and wrote the manuscript; and all authors reviewed and contributed to the preparation of the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: L. Vijaya Mohan Rao, Department of Cellular and Molecular Biology, The University of Texas Health Science Center at Tyler, 11937 US Highway 271, Tyler, TX 75708; e-mail: vijay.rao@uthct.edu.
Requests for original data may be made by contacting the corresponding author, L. Vijaya Mohan Rao (e-mail: vijay.rao@uthct.edu).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal