

Key Points
Inhibition of GSDMD with disulfiram abrogates NET formation, reducing multiple organ dysfunction and sepsis lethality.
Neutrophils from septic patients undergoing NETosis express GSDMD on the cell membrane and around typical NET structures.

Abstract
Multiple organ dysfunction is the most severe outcome of sepsis progression and is highly correlated with a worse prognosis. Excessive neutrophil extracellular traps (NETs) are critical players in the development of organ failure during sepsis. Therefore, interventions targeting NET release would likely effectively prevent NET-based organ injury associated with this disease. Herein, we demonstrate that the pore-forming protein gasdermin D (GSDMD) is active in neutrophils from septic humans and mice and plays a crucial role in NET release. Inhibition of GSDMD with disulfiram or genic deletion abrogated NET formation, reducing multiple organ dysfunction and sepsis lethality. Mechanistically, we demonstrate that during sepsis, activation of the caspase-11/GSDMD pathway controls NET release by neutrophils during sepsis. In summary, our findings uncover a novel therapeutic use for disulfiram and suggest that GSDMD is a therapeutic target to improve sepsis treatment.
Introduction
Sepsis is a life-threatening condition caused by a dysregulated host response to infection.1 Multiple organ dysfunction is the most severe outcome of sepsis and is highly associated with mortality.2,3 Although the mechanisms involved in vital organ damage have not been fully clarified, neutrophil infiltration into organs contributes to tissue damage and the development of organ dysfunction through the release of several cytotoxic mediators, such as free radicals, enzymes, and neutrophil extracellular traps (NETs).4,5
NETs are extracellular DNA matrices containing histone and cytotoxic enzymes, including myeloperoxidase (MPO) and elastase, that are released by neutrophils during NETosis as a part of the arsenal with which neutrophils kill microorganisms.6 However, despite this host-protective role of NETs, increasing evidence has shown that NETs also mediate tissue injury in various inflammatory conditions, including sepsis.7-10 Therefore, interventions to inhibit NETosis would likely be effective in preventing NET-based organ injury associated with sepsis.
Disulfiram was identified in the 19th century, found to inhibit aldehyde dehydrogenase (ALDH) and has been used to treat alcoholism.11 Recently, in addition to inhibiting ALDH, Hu et al12 demonstrated that this drug is a potent inhibitor of gasdermin D (GSDMD) in mouse and human macrophages. GSDMD is a pore-forming protein that acts as a central executioner of inflammatory cell death.13 In macrophages, inflammasome activation by canonical (NLRP4, AIM2, and caspase-1) or noncanonical (caspase-4/5 in humans or caspase-11 in mice) pathways leads to the cleavage of GSDMD, which translocates to the plasma membrane to form pores and induce a lytic proinflammatory form of cell death called pyroptosis.13-15 However, unlike in macrophages, which undergo pyroptosis, inflammasome activation in neutrophils triggers NETosis, which is also a GSDMD-dependent event.16,17 Therefore, the participation of GSDMD in the immunopathology of sepsis is contradictory. Most studies suggest that GSDMD-dependent macrophage pyroptosis plays a harmful role.12,18-20 However, these studies do not exclude a plausible alternative effect of GSDMD mediating NETosis in neutrophils and whether NETs could be the mediator responsible for sepsis multiorgan dysfunction.
Thus, considering that these mechanisms have not been addressed in NETosis-based injury associated with sepsis, in the present study, we investigated whether the inflammasome/GSDMD pathway connected NET release associated with organ injury during polymicrobial sepsis and whether treatment with GSDMD inhibitors, such as disulfiram, could improve sepsis outcomes. GSDMD expression was increased during clinical and experimental sepsis. Additionally, GSDMD inhibition with disulfiram or genic deletion decreased the NET production levels, systemic inflammation, and organ dysfunction, resulting in improved sepsis survival. Mechanistically, the noncanonical caspase-11 inflammasome pathway was required to cleave GSDMD in neutrophils during sepsis. Importantly, neutrophils from patients with sepsis undergoing NETosis express GSDMD on the cell membrane and are associated with typical NET structures. Taken together, these results suggest that GSDMD inhibitors, such as disulfiram, are a potential therapeutic strategy to improve sepsis.
Materials and methods
Patients
Informed consent was received as per the Declaration of Helsinki. Peripheral blood samples were collected from 25 patients with sepsis who were prospectively eligible in the study within the first 24 hours of admission at the Emergency Department of a high-complexity hospital; 24 were finally enrolled. The exclusion criteria included active hematologic malignancy or cancer, chronic treatment with steroids, transplantation, HIV infection, or advanced cirrhosis. Sepsis severity was evaluated using the Sequential Organ Failure Assessment score.21,22 The source of sepsis was pulmonary in 16 cases (66%), urinary infection in 3 (12.5%), abdominal in 2 patients (8.33%), and others in 3 (12.5%). Fifteen patients (62.5%) exhibited severe sepsis, and 9 patients (37.5%) exhibited septic shock. The overall mortality was 8 patients (33.3%). The clinical features of the subjects are detailed in Table 1. Twenty healthy volunteers were included as controls. The study was approved by the Human Subjects Institutional Committee of the Ribeirao Preto Medical School, Brazil (protocol number 134 30459114.6.0000.5440).
Characteristics of the patients with sepsis
| Demographics | n (%) |
|---|---|
| No. | 24 |
| Sex | |
| Female | 12 (50) |
| Male | 12 (50) |
| Age (y), mean ± SEM | 65 ± 3.23 |
| Hospital duration (d), mean ± SEM | 13 ± 3.08 |
| Clinical | |
| Infectious focus | |
| Pulmonary | 16 (66.66) |
| Urinary infection | 3 (12.5) |
| Abdominal | 2 (8.33) |
| Others | 3 (12.5) |
| Charlson Comorbity Index, mean ± SEM | 5 ± 0.45 |
| SOFA, mean ± SEM | 5.5 ± 1.01 |
| APACHE II, mean ± SEM | 21 ± 1.91 |
| Severe sepsis | 15 (62.5) |
| Septic shock | 9 (37.5) |
| Laboratory finding | |
| CRP (mg/dL)* | 15.4 ± 2.13 |
| Hemoglobin (g/dL) | 11.6 ± 0.70 |
| Neutrophils (cell/mm3) | 12 000 ± 1386.97 |
| Lymphocytes (cell/mm3) | 1500 ± 444.65 |
| Platelets (counts/mm3) | 177 500 ± 18 401.98 |
| Outcome | |
| Death | 8 (33.33) |
| Demographics | n (%) |
|---|---|
| No. | 24 |
| Sex | |
| Female | 12 (50) |
| Male | 12 (50) |
| Age (y), mean ± SEM | 65 ± 3.23 |
| Hospital duration (d), mean ± SEM | 13 ± 3.08 |
| Clinical | |
| Infectious focus | |
| Pulmonary | 16 (66.66) |
| Urinary infection | 3 (12.5) |
| Abdominal | 2 (8.33) |
| Others | 3 (12.5) |
| Charlson Comorbity Index, mean ± SEM | 5 ± 0.45 |
| SOFA, mean ± SEM | 5.5 ± 1.01 |
| APACHE II, mean ± SEM | 21 ± 1.91 |
| Severe sepsis | 15 (62.5) |
| Septic shock | 9 (37.5) |
| Laboratory finding | |
| CRP (mg/dL)* | 15.4 ± 2.13 |
| Hemoglobin (g/dL) | 11.6 ± 0.70 |
| Neutrophils (cell/mm3) | 12 000 ± 1386.97 |
| Lymphocytes (cell/mm3) | 1500 ± 444.65 |
| Platelets (counts/mm3) | 177 500 ± 18 401.98 |
| Outcome | |
| Death | 8 (33.33) |
APACHE, acute physiology and chronic health evaluation II; SOFA, sequential organ failure assessment.
CRP, C-reactive protein (normal value <5 mg/dL).
Mice
Casp1−/−, Casp11−/−, Nlrc4−/−, Aim2−/−, and Gsdmd mice in a C57BL/6 J background were obtained from the animal facility of the Ribeirao Preto Medical School of the University of São Paulo, São Paulo, Brazil. The mice were housed in barrier cages under controlled environmental conditions (12/12-hour light/dark cycle; 55 ± 5% humidity; 23°C). The protocol was approved by the Animal Ethics Committee of the Ribeirao Preto Medical School, University of Sao Paulo, Sao Paulo, Brazil (protocol number 149/2019).
Dataset analysis
We used the GEOquery package23 to download author-normalized expression data and sample metadata from the study of Venet et al.24 The limma package was used to identify the differentially expressed genes between patients with septic shock at days 1, 2, or 3 after hospital admission and healthy controls.25 Probes that matched the same gene symbol, according to the annotation of Lima et al,26 were selected by taking the one with the lowest P value. To identify Gene Ontology terms associated with septic shock, we applied the FGSEA package using the log2 fold-change values between septic shock at days 1, 2, or 3 after hospital admission and healthy controls as gene ranks (default parameters).27
Cecal ligation and puncture and endotoxemia models
A cecal ligation and puncture model (CLP) was performed as previously described.8 The mice were briefly anesthetized (isoflurane 1% to 3%), and 2 punctures were made through the cecum using an 18-gauge needle to induce severe CLP sepsis. Control mice (sham) were submitted to the same procedures without cecal puncture. Previously, it was demonstrated that the levels of inflammatory markers and organ lesions in sham mice are not different from those in naïve mice.28,29 All mice received 1 mL saline subcutaneous(ly) (SC) immediately after surgery. Survival was observed for up to 7 days. In the endotoxemia model, the mice received a single lipopolysaccharide (LPS) injection (10 mg/kg, intraperitoneal[ly] [IP]).
Treatment with GSDMD inhibitor (disulfiram)
The mice were injected with vehicle (dimethyl sulfoxide, 1% v/v) or disulfiram (Sanofi-Aventis South America) at 80 mg/kg SC at 24 and 4 hours before CLP or LPS injection. Subsequently, the animals were treated for 6 hours and then every 12 hours for 2 days for survival rate analysis as previously reported.12
Neutrophil preparation and stimulation
Mouse bone marrow neutrophils and human circulating neutrophils were also isolated using Percoll density gradients. Briefly, 2 gradients (72% and 65%) were prepared to isolate mouse bone marrow neutrophils, and 4 different gradients, 72%, 65%, 54%, and 45%, were used to isolate human circulating neutrophils. After centrifugation at 600g for 30 minutes at 4°C, the cell layer at the 72% gradient interface was collected as the neutrophil fraction. To investigate inflammasome activation in neutrophils, we reproduced a classical stimulus used in macrophages and neutrophils. Briefly, neutrophils were primed with Pam3CSK4 (1 μg/mL; InvivoGen) to suppress basal neutrophil apoptosis and induce the expression of caspase-11, NLRP3, and pro-interleukin (IL)-1β and then were transfected with ultrapure LPS (10 μg/mL; Sigma) into the cytosol with DOTAP Liposomal Transfection Reagent (Roche) in RPMI (Gibco) to activate caspase-11. Phorbol myristate acetate (50 nM; Sigma) or ionomycin (5 μM; Sigma) was used as classical NET stimuli for subsequent assessments.17,30 In another experiment, neutrophils were treated with disulfiram (GSDMD inhibitor, 30 μM), sivelestat (neutrophil elastase inhibitor, 10 μM), or vehicle (dimethyl sulfoxide, 1% v/v) 1 hour before stimulation as described above.
NET quantification (MPO/DNA assay)
This procedure was performed as previously described.31,32 Briefly, an antibody bound to a 96-well clear-bottomed black plate captured the enzyme MPO (5 μg/mL; Abcam). According to the manufacturer's instructions, the amount of DNA bound to the enzyme was quantified using the Quant-iT PicoGreen Kit (Invitrogen). The fluorescence intensity (excitation at 488 nm and emission at 525 nm) was quantified using a FlexStation 3 Microplate Reader (Molecular Devices).
Adoptive cell transfer
Mouse bone marrow neutrophils and monocytes from 6- to 8-week-old C57/BL6 mice were isolated. Using Percoll density gradients, we obtained neutrophils and peripheral blood mononuclear cell. To further enrich the monocyte population from peripheral blood mononuclear cells, Ly6C+ cells were isolated using single-stained anti-phycoerythrin capture magnetic beads. The cells were then stained with 5 µM CellTrace dye according to the manufacturer’s instructions (Invitrogen). We introduced 1 × 107 cells per mouse in a volume of 50 μL sterile phosphate-buffered saline (PBS) by IV injection into the ophthalmic vein33 of Gsdmd−/− mice. Next, 1 hour after cell transfer, we performed CLP, as previously described. Twelve hours after CLP, blood samples and spleens were collected to analyze organ injury markers, cytokines, and viable transfer neutrophils and monocytes.
Blood biomarkers of organ injury analysis
The animals were euthanized 6, 12, and 24 hours after CLP or 24 hours after LPS injection, and the plasma samples were collected to measure renal, hepatic, and cardiac dysfunctions, as assessed by blood urea nitrogen (BUN), aspartate transaminase (AST), alanine transaminase (ALT), creatine kinase-MB isoenzyme (CK-MB), and troponin I, respectively. The assays were performed according to the manufacturer's instructions (Labtest and Elabsience).
Cytokine assays (enzyme-linked immunosorbent assay)
According to the manufacturer's instructions, the cytokine concentrations were measured in the plasma by enzyme-linked immunosorbent assay using antibodies from R&D Systems. The optical density of the individual samples was measured at 450 nm using a spectrophotometer (Spectra Max-250; Molecular Devices).
Histologic examination
The mice were euthanized 24 hours after sepsis. The lung tissue was harvested and fixed in 4% buffered formalin and embedded in paraffin blocks. Sections (5 μm) were then stained with hematoxylin and eosin for histologic examination. Images were acquired using a DMI 6000B microscope (Leica Microsystems) at ×200 or ×400 magnification.
Immunofluorescence staining and confocal microscopy
Neutrophils were attached to slides coated with a poly-l-lysine (0.1%) solution (Sigma) for 4 hours and then fixed with paraformaldehyde (4%). The samples were washed with PBS and blocked with 1% bovine serum albumin and 22.52 mg/mL glycine in PBST (PBS + 0.1% Tween 20). The slides were stained with the following antibodies: (1) rabbit anti-histone H3 (H3Cit; 1:500, #ab5103; Abcam); (2) mouse anti-MPO (2C7; Abcam cat. ab25989; 1:500); and (3) rabbit anti-GSDMD (#ab57785; Abcam). Next, the samples were incubated with donkey anti-mouse immunoglobulin G Alexa Fluor 488 (1:800, #150061; Abcam) and donkey anti-rabbit immunoglobulin G Alexa Fluor 594 (1:800, #ab150076; Abcam) secondary antibodies. The nuclei were stained with 4',6-diamidino-2-phenylindole dihydrochloride (DAPI; 1:1.000, D1306; Life Technologies). Images were acquired using Axio Observer combined with an LSM 780 confocal microscope system at 630× magnification (Carl Zeiss). All acquired images were analyzed using Fiji by ImageJ.
Statistical analysis
The data were reported as the means ± standard error of the mean (SEM) of the values obtained from at least 2 independent experiments. The means of different treatments were compared by Student t test or 1-way analysis of variance (ANOVA), followed by a Tukey’s test as appropriate. The survival rate was expressed as the percentage of live animals, and the Mantel-Cox log-rank test was used to determine the differences between survival curves. A value of P < .05 was considered significant. All statistical analyses were performed using GraphPad Prism version 8.0.1 for Windows (GraphPad Software).
Results
Gasdermin D deficiency prevents NET release and reduces organ dysfunction during polymicrobial sepsis
To determine whether GSDMD is involved in NET release and the physiopathology of sepsis, we induced sepsis by CLP in GSDMD-deficient (Gsdmd−/−) and wild-type (WT) control mice. As shown in Figure 1A, CLP-induced sepsis stimulated robust and sustained intravascular release of NETs in WT mice, with a peak at 12 hours after surgery. Notably, the release of NETs was abrogated in Gsdmd−/− mice. To confirm whether GSDMD activation is involved in NET generation, we stimulated primed neutrophils with cytosolic LPS as previously described.17,30 Consistent with the in vivo findings, cytosolic LPS triggered NET production in WT neutrophils but not in Gsdmd−/− neutrophils (Figure 1B-C). Additionally, we observed that NET induction by phorbol myristate acetate (PMA) - reactive oxygen species dependent- and ionomycin (calcium ionophore) was also prevented in Gsdmd−/− neutrophils (supplemental Figure 1A-B available on the Blood Web site). Next, given that the concentration of NETs has been positively correlated with multiorgan dysfunction and the severity of sepsis,8,22,23 we investigated the role of GSDMD in septic organ dysfunction. Gsdmd−/− mice showed lower plasma levels of organ injury markers, including cardiac CK-MB and troponin-I (Figure 1D; Supplemental Figure 1C), renal BUN (Figure 1E), and hepatic ALT and AST (supplemental Figure 1D-E) than WT mice. Similarly, GSDMD deficiency prevented the development of the systemic inflammatory response characterized by low plasma concentrations of tumor necrosis factor α (TNF-α), IL-1β, and IL-6, which were observed at high levels in septic WT mice (Figure 1F-G; supplemental Figure 1F). Histopathologic analysis of the lung tissue revealed enhanced pulmonary vascular congestion, edema, and significant neutrophil infiltration in septic WT mice but not in Gsdmd−/− mice (Figure 1H). To assess cardiovascular dysfunctions, hemodynamic parameters were monitored continuously for 24 hours after CLP using implantable telemetry devices. Sepsis led to marked reductions in the mean arterial pressure (MAP) and heart rate (HR), particularly at 24 hours after CLP induction in WT mice, and these effects were prevented in Gsdmd−/− mice (Figure 1I; supplemental Figure 1G). Finally, an improvement in the survival rate of Gsdmd−/− mice was observed compared with that of WT mice in the context of CLP-induced sepsis (80% vs 20%, respectively; Figure 1J). Taken together, these results indicate that GSDMD participates in NET production and consequent systemic inflammation, organ failure, and mortality during sepsis. That the peak plasma concentration of NETs preceded the highest concentrations of organ damage markers supports the hypothesis that NETs mediate multiorgan damage.
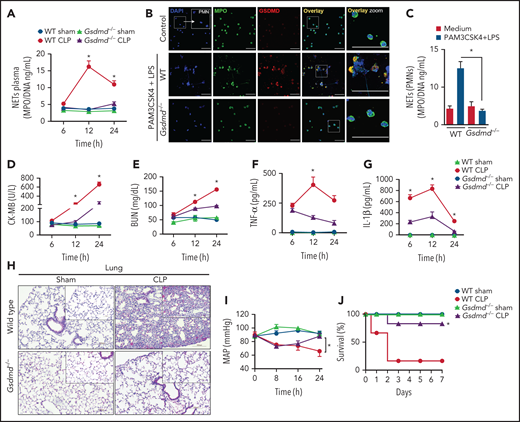
Gasdermin D contributes to organ dysfunction in sepsis by NET release. (A) The MPO/DNA-NET concentrations in the plasma from WT and Gsdmd−/− mice were determined at 6, 12, or 24 hours after sepsis induction by CLP. (B) Bone marrow neutrophils from WT and Gsdmd−/− mice were primed with PAM3CSK4 (1 µg/mL) for 4 hours and then transfected with ultrapure LPS (10 µg/mL) for 4 hours. Representative fluorescence images of NETs stained for DNA (DAPI, blue), myeloperoxidase (MPO, green), and the gasdermin D cleaved fraction (GSDMD, red) are shown. Scale bar, 50 µm at ×630 magnification. (C) The concentrations of MPO/DNA-NETs in the neutrophil culture supernatants after 4 hours of stimulation were determined using the picogreen test. (D-E) The plasma levels of the organ injury markers CK-MB and BUN and (F-G) TNF-α and IL-1β were determined at 6, 12, and 24 hours after sepsis induction by CLP. (H) Representative histopathology images of lung tissue sections 24 hours after sepsis induction by CLP are shown at ×200 magnification. The square insets represent the image at ×400 magnification. (I) Blood pressure was continuously monitored by telemetry, which was used to calculate the MAP for 24 hours after sepsis induction by CLP. (J) WT and Gsdmd−/− mice were subjected or not to CLP, and survival was recorded for 7 days. The data are expressed as means ± SEM. *P < .05; WT CLP vs Gsdmd−/− CLP, Student t test (A,D-I), 1-way ANOVA followed by Tukey's test (C); H-Mantel-Cox log-rank test (J). The data are representative of ≥2 independent experiments, each including 5-7 animals per group.
Gasdermin D contributes to organ dysfunction in sepsis by NET release. (A) The MPO/DNA-NET concentrations in the plasma from WT and Gsdmd−/− mice were determined at 6, 12, or 24 hours after sepsis induction by CLP. (B) Bone marrow neutrophils from WT and Gsdmd−/− mice were primed with PAM3CSK4 (1 µg/mL) for 4 hours and then transfected with ultrapure LPS (10 µg/mL) for 4 hours. Representative fluorescence images of NETs stained for DNA (DAPI, blue), myeloperoxidase (MPO, green), and the gasdermin D cleaved fraction (GSDMD, red) are shown. Scale bar, 50 µm at ×630 magnification. (C) The concentrations of MPO/DNA-NETs in the neutrophil culture supernatants after 4 hours of stimulation were determined using the picogreen test. (D-E) The plasma levels of the organ injury markers CK-MB and BUN and (F-G) TNF-α and IL-1β were determined at 6, 12, and 24 hours after sepsis induction by CLP. (H) Representative histopathology images of lung tissue sections 24 hours after sepsis induction by CLP are shown at ×200 magnification. The square insets represent the image at ×400 magnification. (I) Blood pressure was continuously monitored by telemetry, which was used to calculate the MAP for 24 hours after sepsis induction by CLP. (J) WT and Gsdmd−/− mice were subjected or not to CLP, and survival was recorded for 7 days. The data are expressed as means ± SEM. *P < .05; WT CLP vs Gsdmd−/− CLP, Student t test (A,D-I), 1-way ANOVA followed by Tukey's test (C); H-Mantel-Cox log-rank test (J). The data are representative of ≥2 independent experiments, each including 5-7 animals per group.
Gasdermin D in neutrophils contributes to sepsis-induced organ dysfunction
Because Gsdmd−/− mice are deficient in GSDMD in all immune cells, we investigated whether GSDMD activation in neutrophils is relevant to the physiopathology of sepsis. Thus, we performed a cell transfer experiment in which the same number of neutrophils or monocytes (1 × 107 cells per mouse) from WT mice was transferred to Gsdmd−/− mice. Next, the animals were subjected to CLP, and the levels of NETs, vital organ injury markers, and inflammatory cytokines were evaluated 12 hours after CLP (Figure 2A). The injection of WT neutrophils, but not monocytes, into Gsdmd−/− mice restored NET production (Figure 2B). Additionally, increased levels of organ injury markers (CK-MB, BUN, ALT, and AST; Figure 2C-F) and inflammatory cytokines (TNF-α, IL-6, and IL-1β; Figure 2G-I) were observed in the serum of Gsdmd−/− mice that received WT neutrophils. Although the mice that received monocytes from WT animals did not present a significant increase in organ injury markers compared with those in Gsdmd−/− CLP-induced mice, the observed levels of cytokines (IL-6 and IL-1β) increased significantly compared with those observed in Gsdmd−/− animals (Figure 2H-I). Notably, the presence of viable neutrophils and monocytes was observed 12 hours after cell transfer in the circulation or spleen of the transplanted animals (supplemental Figure 2). Taken together, these data suggest that GSDMD in neutrophils is a key factor in the pathophysiology of organ dysfunction associated with sepsis.
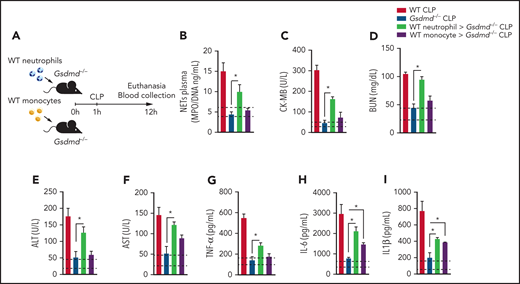
Gasdermin D expression in neutrophils is essential for organ dysfunction during polymicrobial sepsis. (A) Schematic representation of the cell transfer experiment in which neutrophils (1 × 107 cells per mouse) or monocytes (1 × 107 cells per mouse) from WT animals were transferred to Gsdmd−/− mice via IV injection 1 hour after the mice were subjected to CLP and 12 hours after the animals were euthanized. (B) Circulating concentrations of MPO/DNA-NETs were quantified 12 hours after sepsis induction by CLP. (C-F) The plasma levels of organ injury markers and (G-I) cytokines TNF-α, IL-6, and IL-1β were determined 12 hours after sepsis induction by CLP. (B-I) The dashed lines indicate the upper and lower levels of the mediators measured in the control sham groups. The data are expressed as means ± SEM. *P < .05; 1-way ANOVA followed by Tukey’s test (B-I). The data are representative of ≥2 independent experiments, each including 4 to 6 animals per group.
Gasdermin D expression in neutrophils is essential for organ dysfunction during polymicrobial sepsis. (A) Schematic representation of the cell transfer experiment in which neutrophils (1 × 107 cells per mouse) or monocytes (1 × 107 cells per mouse) from WT animals were transferred to Gsdmd−/− mice via IV injection 1 hour after the mice were subjected to CLP and 12 hours after the animals were euthanized. (B) Circulating concentrations of MPO/DNA-NETs were quantified 12 hours after sepsis induction by CLP. (C-F) The plasma levels of organ injury markers and (G-I) cytokines TNF-α, IL-6, and IL-1β were determined 12 hours after sepsis induction by CLP. (B-I) The dashed lines indicate the upper and lower levels of the mediators measured in the control sham groups. The data are expressed as means ± SEM. *P < .05; 1-way ANOVA followed by Tukey’s test (B-I). The data are representative of ≥2 independent experiments, each including 4 to 6 animals per group.
The caspase-11/gasdermin D pathway plays a key role in NET release and organ dysfunction during sepsis
To further explore the mechanism of GSDMD activation in neutrophils during sepsis, we performed experiments using Aim2−/−, Nlrc4, Casp1−/−, and Casp11−/− mice, all GSDMD upstream inflammasome-related genes, to determine which of them mimic sepsis resistance observed in Gsdmd−/− mice. Strikingly, Casp11−/− mice, but not Aim2−/−, Nlrc4, or Casp1−/− mice, showed significant improvements in the survival rates after sepsis induction compared with WT septic mice, suggesting that the noncanonical inflammasome pathway mediated by caspase-11 is involved in GSDMD activation and NET release during sepsis (Figure 3A-D). To confirm this hypothesis, primed neutrophils from WT and Casp11−/− mice were stimulated with cytosolic LPS as previously described.17,30 We observed that caspase-11 was required to trigger GSDMD cleavage, a finding that was confirmed by reduced expression of the N-terminal fragment of GSDMD (GSDMD-N; Figure 3E-F). By contrast, the absence of GSDMD did not interfere with caspase-11 activation, suggesting that caspase-11 is upstream of GSDMD (supplemental Figure 3). Additionally, unlike control neutrophils, neutrophils harvested from Casp11−/− mice could not produce NETs after cytosolic LPS stimulation (Figure 3G-H). Thus, caspase-11 is likely implicated in GSDMD activation during sepsis, which is involved in NET release. Accordingly, the Casp11−/− septic mice showed reduced circulating levels of NETs after sepsis induction (Figure 4A). This change in NETs was accompanied by reductions in the biochemical markers of organ damage CK-MB, troponin-I, BUN, ALT, and AST (Figure 4B-F) and attenuation of the severity of histopathologic changes in lung tissue, such as vascular congestion, neutrophil infiltration, and edema, in septic Casp11−/− mice compared with those in the WT group (Figure 4G). Furthermore, significant reductions in the plasma concentrations of the cytokines TNF-α, IL-6, and IL-1β were observed (Figure 4H-J). We also assessed the hemodynamic parameters of septic WT and Casp11−/− mice. The MAP and HR were reduced progressively during the first 24 hours after CLP in WT septic mice, and these reductions did not occur in Casp11−/− mice (Figure 4K-L). Taken together, these data suggest that GSDMD activation via caspase-11 mediates NET release, leading to organ failure during sepsis. However, caspase-11 deletion does not completely prevent the release of NETs during sepsis (Figure 4A). This result suggests that caspase-independent mechanisms may be involved in GSDMD activation and NET formation during sepsis. In neutrophils, GSDMD can also be cleaved by serine proteases, such as neutrophil elastase (NE).34 Consistent with this finding, we observed that NE inhibition reduced GSDMD activation and NETosis after stimulation with cytosolic LPS (supplemental Figure 4). In accordance, other studies that use the NE inhibitor sivelestat in similar conditions also observed reduction of NETs.35,36 These results suggest that NE is an alternative mechanism of GSDMD activation and NET induction under infectious stimuli.
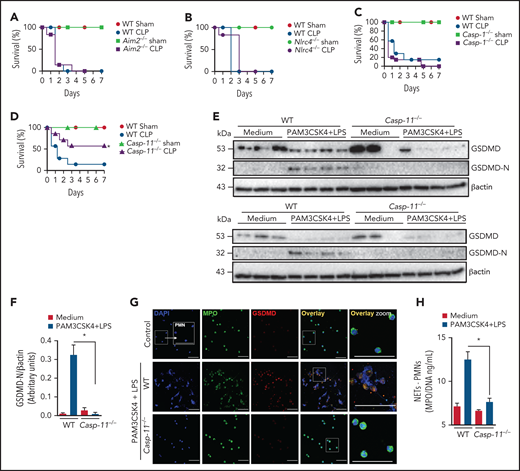
Caspase-11 contributes to NET release through gasdermin D. (A-D) The survival of WT, Aim2−/−, Nlrc4−/−, Casp1−/−, and Casp11−/− mice subjected or not to CLP was followed for 7 days. (E) Bone marrow neutrophils from WT and Casp11−/− mice were primed with PAM3CSK4 (1 μg/mL) for 4 hours and then transfected with ultrapure LPS (10 μg/mL) for 4 hours. The cell lysates were harvested for immunoblot analysis of GSDMD total and its cleaved fraction (GSDMD-N). Each band represents 1 different animal. Actin (β-actin) was used as a loading control. (F) Quantification of GSDMD-N relative to β-actin. Bone marrow neutrophils from WT and Casp11−/− mice were primed with PAM3CSK4 (1 µg/mL) for 4 hours and then transfected with ultrapure LPS (10 µg/mL) for 4 hours. (G) Representative fluorescence images of NETs stained for DNA (DAPI, blue), myeloperoxidase (MPO, green), and the gasdermin D cleaved fraction (GSDMD, red) are shown. Scale bar, 50 µm at ×630 magnification. (H) The concentration of MPO/DNA-NETs was determined in the culture supernatants. The data are expressed as means ± SEM. *P < .05; H-Mantel-Cox log-rank test (A-D), 1-way ANOVA followed by Tukey’s test (F,H). The data are representative of ≥2 independent experiments, each including 5 to 7 animals per group.
Caspase-11 contributes to NET release through gasdermin D. (A-D) The survival of WT, Aim2−/−, Nlrc4−/−, Casp1−/−, and Casp11−/− mice subjected or not to CLP was followed for 7 days. (E) Bone marrow neutrophils from WT and Casp11−/− mice were primed with PAM3CSK4 (1 μg/mL) for 4 hours and then transfected with ultrapure LPS (10 μg/mL) for 4 hours. The cell lysates were harvested for immunoblot analysis of GSDMD total and its cleaved fraction (GSDMD-N). Each band represents 1 different animal. Actin (β-actin) was used as a loading control. (F) Quantification of GSDMD-N relative to β-actin. Bone marrow neutrophils from WT and Casp11−/− mice were primed with PAM3CSK4 (1 µg/mL) for 4 hours and then transfected with ultrapure LPS (10 µg/mL) for 4 hours. (G) Representative fluorescence images of NETs stained for DNA (DAPI, blue), myeloperoxidase (MPO, green), and the gasdermin D cleaved fraction (GSDMD, red) are shown. Scale bar, 50 µm at ×630 magnification. (H) The concentration of MPO/DNA-NETs was determined in the culture supernatants. The data are expressed as means ± SEM. *P < .05; H-Mantel-Cox log-rank test (A-D), 1-way ANOVA followed by Tukey’s test (F,H). The data are representative of ≥2 independent experiments, each including 5 to 7 animals per group.
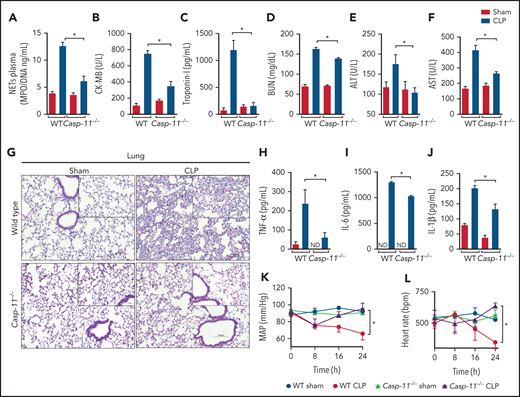
Caspase-11 deletion prevents NET release and organ dysfunction during polymicrobial sepsis. (A) Circulating amounts of MPO/DNA-NETs were quantified 24 hours after sepsis induction by CLP. (B-F) Plasma levels of organ injury markers. (G) Representative images of H&E staining of lung tissue sections from WT and Casp11−/− mice 24 hours after sepsis induction by CLP are shown at ×200 magnification. The square insets represent the image at ×400 magnification. (H-J) The plasma levels of the cytokines TNF-α, IL-6, and IL-1β (G-I) were determined 24 hours after sepsis induction by CLP. (K-L) WT and Casp11−/− mice were implanted using a telemetric pressure transmitter probe to determine the MAP and HR 24 hours after CLP-induced sepsis. The data are expressed as means ± SEM. *P < .05; 1-way ANOVA followed by Tukey’s test (A-F,H-J), CLP WT vs CLP Casp11−/− Student t test (K,L). The data are representative of ≥ 2 independent experiments, each including 4 to 7 animals per group.
Caspase-11 deletion prevents NET release and organ dysfunction during polymicrobial sepsis. (A) Circulating amounts of MPO/DNA-NETs were quantified 24 hours after sepsis induction by CLP. (B-F) Plasma levels of organ injury markers. (G) Representative images of H&E staining of lung tissue sections from WT and Casp11−/− mice 24 hours after sepsis induction by CLP are shown at ×200 magnification. The square insets represent the image at ×400 magnification. (H-J) The plasma levels of the cytokines TNF-α, IL-6, and IL-1β (G-I) were determined 24 hours after sepsis induction by CLP. (K-L) WT and Casp11−/− mice were implanted using a telemetric pressure transmitter probe to determine the MAP and HR 24 hours after CLP-induced sepsis. The data are expressed as means ± SEM. *P < .05; 1-way ANOVA followed by Tukey’s test (A-F,H-J), CLP WT vs CLP Casp11−/− Student t test (K,L). The data are representative of ≥ 2 independent experiments, each including 4 to 7 animals per group.
Pharmacologic inhibition of gasdermin D prevents NET release and organ dysfunction in polymicrobial sepsis
A recent study showed that disulfiram, a drug used to treat alcohol addiction,37 potently inhibits GSDMD inflammasome-mediated pyroptosis.12 Herein, we investigated whether the pharmacologic inhibition of GSDMD with disulfiram prevented NETosis and consequent organ dysfunction during polymicrobial sepsis. We observed that primed neutrophils treated with disulfiram did not produce NETs after cytosolic LPS stimulation (Figure 5A). Additionally, disulfiram reduced the processing of histone H3, an effect also observed in Casp-11−/− and Gsdmd−/− neutrophils (supplemental Figure 5). Next, WT septic mice were treated with vehicle or disulfiram.12 The circulating levels of NETs were reduced by disulfiram compared with vehicle treatment (Figure 5B). The plasma concentrations of biochemical organ injury markers, including CK-MB (heart), BUN (kidney), and ALT and AST (liver), were also reduced (Figure 5C-F). Histologic analysis showed reduced edema and vascular congestion in the lung tissue of mice treated with the GSDMD inhibitor (Figure 5G). The levels of systemic inflammatory cytokines (TNF-α, IL-6, and IL-1β) were mitigated in the septic disulfiram-treated group compared with that in the control group (Figure 5H-J). Furthermore, similar to those of Gsdmd−/− mice, treatment with disulfiram improved the survival rates of animals subjected to polymicrobial sepsis (Figure 5K). We also observed that GSDMD inhibition with disulfiram protected mice from endotoxemia induced by LPS. We observed reductions in the plasma levels of NETs (supplemental Figure 6A), organ injury markers (CK-MB, BUN, ALT, and AST; supplemental Figure 6B-E), and lung tissue damage (supplemental Figure 6F) and an improved overall survival rate in the disulfiram-treated group compared with that in the vehicle control group (supplemental Figure 6G). Furthermore, disulfiram did not substantially affect the serum levels of NETs (supplemental Figure 7A), inflammatory cytokines (TNF-α, IL-6, and IL-1β; supplemental Figure 7B-D), lung tissue damage (supplemental Figure 7E), organ lesion markers (CK-MB, BUN, ALT, and AST; supplemental Figure 6F-I) or overall survival (supplemental Figure 7J) in Gsdmd−/− mice, reinforcing that the protective action of disulfiram in sepsis is dependent on GSDMD. Thus, these findings suggest that pharmacologic inhibition of GSDMD with disulfiram prevents NET release and organ dysfunction, improving sepsis outcomes.
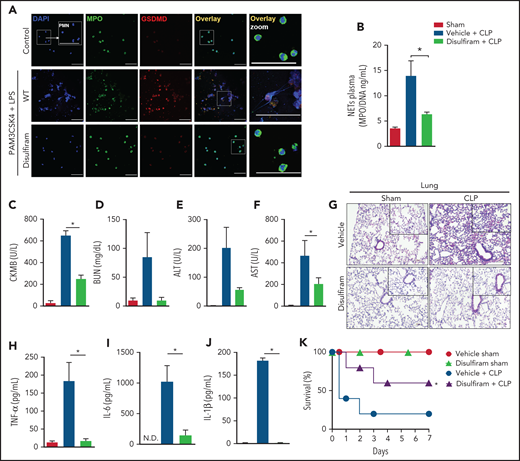
Disulfiram, a gasdermin D inhibitor, protects against CLP-induced sepsis by reducing NET release. (A) Bone marrow neutrophils were treated with disulfiram (30 µM) or vehicle 1 hour before stimulation with PAM3CSK4 (1 µg/mL) for 4 hours and then were transfected with ultrapure LPS (10 µg/mL) for an additional 4 hours. Representative fluorescence images of NETs stained for DNA (DAPI, blue), myeloperoxidase (MPO, green), and the gasdermin D cleaved fraction (GSDMD, red) are shown. Scale bar, 50 µm at ×630 magnification. (B) The mice were pretreated with disulfiram (80 mg/kg) or vehicle by subcutaneous injection 24 and 4 hours before surgery and at 6 and 18 hours after CLP. The MPO/DNA-NET concentration in the plasma was determined 24 hours after CLP-induced sepsis. (C-F) The circulating levels of organ injury markers were determined 24 hours after sepsis induction by CLP. (G) Representative images of the histologic staining of the lung sections performed 24 hours after sepsis induction are shown. Scale bar 200 μm at ×400 magnification. (H-J) The systemic levels of TNF-α, IL-6, and IL-1β were measured by enzyme-linked immunosorbent assay 24 hours after CLP-induced sepsis. (K) The mice were pretreated with disulfiram (80 mg/kg) or vehicle by SC injection 24 and 4 hours before CLP-induced sepsis, 6 hours after CLP, and every 12 hours for 2 days and then were followed for survival analysis. The data are expressed as means ± SEM. *P < .05; 1-way ANOVA followed by Tukey’s test (B-F,H-J), H-Mantel-Cox log-rank test (K). The data are representative of ≥2 independent experiments, each including 5 to 7 animals per group.
Disulfiram, a gasdermin D inhibitor, protects against CLP-induced sepsis by reducing NET release. (A) Bone marrow neutrophils were treated with disulfiram (30 µM) or vehicle 1 hour before stimulation with PAM3CSK4 (1 µg/mL) for 4 hours and then were transfected with ultrapure LPS (10 µg/mL) for an additional 4 hours. Representative fluorescence images of NETs stained for DNA (DAPI, blue), myeloperoxidase (MPO, green), and the gasdermin D cleaved fraction (GSDMD, red) are shown. Scale bar, 50 µm at ×630 magnification. (B) The mice were pretreated with disulfiram (80 mg/kg) or vehicle by subcutaneous injection 24 and 4 hours before surgery and at 6 and 18 hours after CLP. The MPO/DNA-NET concentration in the plasma was determined 24 hours after CLP-induced sepsis. (C-F) The circulating levels of organ injury markers were determined 24 hours after sepsis induction by CLP. (G) Representative images of the histologic staining of the lung sections performed 24 hours after sepsis induction are shown. Scale bar 200 μm at ×400 magnification. (H-J) The systemic levels of TNF-α, IL-6, and IL-1β were measured by enzyme-linked immunosorbent assay 24 hours after CLP-induced sepsis. (K) The mice were pretreated with disulfiram (80 mg/kg) or vehicle by SC injection 24 and 4 hours before CLP-induced sepsis, 6 hours after CLP, and every 12 hours for 2 days and then were followed for survival analysis. The data are expressed as means ± SEM. *P < .05; 1-way ANOVA followed by Tukey’s test (B-F,H-J), H-Mantel-Cox log-rank test (K). The data are representative of ≥2 independent experiments, each including 5 to 7 animals per group.
Gasdermin D is associated with NET release in patients with sepsis
To demonstrate the clinical relevance of our findings, we analyzed the transcriptome data of whole blood samples from patients with sepsis and healthy volunteers (Gene Expression Omnibus database; accession number GSE95233). We observed that neutrophil degranulation, neutrophil-mediated immunity, and neutrophil activation involved in the immune response were the top 3 gene sets that were most highly associated with no surviving patients with sepsis at day 1 after intensive care unit (ICU) admission compared with healthy volunteers (Figure 6A). Furthermore, Gsdmd and inflammasome-related genes (Aim2, Nlrc4, Casp1, and Casp5/Casp-11) were upregulated in leukocytes from patients with sepsis compared with those from healthy volunteers (Figure 6B-C). Next, we enrolled 24 patients with severe sepsis or septic shock and 20 healthy volunteers as controls. The demographic and clinical characteristics of septic patients are shown in Table 1 and described in the “Methods” section. First, we confirmed that septic patients showed higher levels of circulating NETs than healthy volunteers (Figure 6D), a finding that was consistent with our findings in the mouse model. Using confocal microscopy, we showed that neutrophils isolated from septic patients produced NETs and expressed GSDMD (Figure 6E [white arrow],F). Furthermore, GSDMD accumulated on the cell membrane and in typical NET structures containing extracellular DNA (Figure 6G, white arrow). Additionally, NET production by blood neutrophils isolated from patients with sepsis was abrogated by GSDMD inhibition with disulfiram (Figure 6H-I). Collectively, these results indicate that the inflammasome/GSDMD pathway is involved in the release of NETs during sepsis.
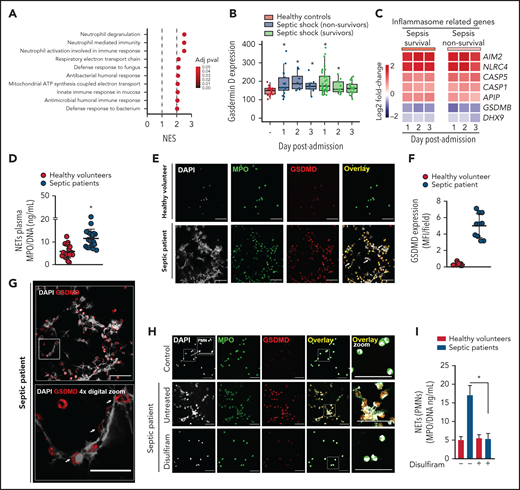
Neutrophils from septic patients who undergo NETosis express gasdermin D. (A) Gene set enrichment analysis identified “neutrophil degranulation,” “neutrophil-mediated immunity,” and “neutrophil activation involved in immune response gene sets” as the top 3 GO terms with the highest normalized enrichment score in nonsurvivor patients with septic shock at day 1 after ICU admission compared with healthy volunteers (adjusted P < .1). (B) Expression levels of Gsdmd in healthy volunteers and patients with septic shock (adjusted P < .1). (C) Heatmap representation of differentially expressed genes in surviving and nonsurviving patients with septic shock compared with healthy volunteers at days 1, 2, and 3 after ICU admission (adjusted P < .1). (D) Circulating amounts of MPO/DNA-NETs were quantified from the plasma of patients with sepsis and healthy volunteers. (E) Neutrophils from patients with sepsis and healthy volunteers were isolated and cultured for 4 hours at 37°C. Representative immunostaining images for DNA (DAPI, gray), myeloperoxidase (MPO, green), and the gasdermin D cleaved fraction (GSDMD, red) are shown. Scale bar, 50 µm at ×630 magnification. (F) GSDMD expression was quantified by Median Fluorescence Intensity (MFI) per field. (G) Magnified fluorescence microscopy images of panel E for DNA (DAPI, gray) and the gasdermin D cleaved fraction (GSDMD, red) are shown. Scale bar, 50 µm at ×630 magnification. Furthermore, to demonstrate the details of GSDMD in neutrophil membranes and its association with NETs (G, top), we performed a 4× digital zoom of the inset square (G, bottom). (H) Neutrophils from patients with sepsis and healthy volunteers were isolated, treated with disulfiram (30 µM) or vehicle, and cultured for 4 hours at 37°C. Representative fluorescence images of NETs stained for DNA (DAPI, gray), myeloperoxidase (MPO, green), and the gasdermin D cleaved fraction (GSDMD, red) are shown. Scale bar, 50 µm at ×630 magnification. (I) The concentrations of MPO/DNA-NETs in the neutrophil culture supernatant after 4 hours were determined using the picogreen test. The data are expressed as means ± SEM. *P < .05; Student t test (D), 1-way ANOVA, followed by Tukey’s test (I). The data are representative of ≥2 independent experiments.
Neutrophils from septic patients who undergo NETosis express gasdermin D. (A) Gene set enrichment analysis identified “neutrophil degranulation,” “neutrophil-mediated immunity,” and “neutrophil activation involved in immune response gene sets” as the top 3 GO terms with the highest normalized enrichment score in nonsurvivor patients with septic shock at day 1 after ICU admission compared with healthy volunteers (adjusted P < .1). (B) Expression levels of Gsdmd in healthy volunteers and patients with septic shock (adjusted P < .1). (C) Heatmap representation of differentially expressed genes in surviving and nonsurviving patients with septic shock compared with healthy volunteers at days 1, 2, and 3 after ICU admission (adjusted P < .1). (D) Circulating amounts of MPO/DNA-NETs were quantified from the plasma of patients with sepsis and healthy volunteers. (E) Neutrophils from patients with sepsis and healthy volunteers were isolated and cultured for 4 hours at 37°C. Representative immunostaining images for DNA (DAPI, gray), myeloperoxidase (MPO, green), and the gasdermin D cleaved fraction (GSDMD, red) are shown. Scale bar, 50 µm at ×630 magnification. (F) GSDMD expression was quantified by Median Fluorescence Intensity (MFI) per field. (G) Magnified fluorescence microscopy images of panel E for DNA (DAPI, gray) and the gasdermin D cleaved fraction (GSDMD, red) are shown. Scale bar, 50 µm at ×630 magnification. Furthermore, to demonstrate the details of GSDMD in neutrophil membranes and its association with NETs (G, top), we performed a 4× digital zoom of the inset square (G, bottom). (H) Neutrophils from patients with sepsis and healthy volunteers were isolated, treated with disulfiram (30 µM) or vehicle, and cultured for 4 hours at 37°C. Representative fluorescence images of NETs stained for DNA (DAPI, gray), myeloperoxidase (MPO, green), and the gasdermin D cleaved fraction (GSDMD, red) are shown. Scale bar, 50 µm at ×630 magnification. (I) The concentrations of MPO/DNA-NETs in the neutrophil culture supernatant after 4 hours were determined using the picogreen test. The data are expressed as means ± SEM. *P < .05; Student t test (D), 1-way ANOVA, followed by Tukey’s test (I). The data are representative of ≥2 independent experiments.
Discussion
This report demonstrated that the pore-forming protein GSDMD controls NET extrusion and subsequent development of organ dysfunction during sepsis. Using polymicrobial or LPS sepsis models, genetic knockout or pharmacologic inhibition of GSDMD with disulfiram prevented NET release and reduced organ lesions, increasing the survival rates of septic mice. Importantly, we confirmed that neutrophils from septic patients undergoing NETosis expressed GSDMD on cell membranes and in association with typical NET structures.
The role of NETs in controlling infection was first described by Brinkmann et al.6 However, over the last decade, we and others have reported that NETs acts as a double-edged sword. In addition to microbicidal activity, NETs also exert toxic effects on host tissues in different diseases, such as rheumatoid arthritis, diabetes, and sepsis.38-42 In this context, NET-associated histones play a role in cytotoxicity because of their ability to modify cell membrane integrity.43-45 Other NET constituents can also contribute to cytotoxicity, such as defensins and elastases, which permeabilize cell membranes and perturb the integrity of cell junctions, respectively.46,47 Our results demonstrated that NET extrusion during sepsis is a GSDMD-dependent event.
GSDMD activation during sepsis is not fully understood. Previous reports have suggested that GSDMD-dependent pyroptosis in macrophages plays a role in the physiopathology of sepsis induced by LPS.12,18 However, these studies do not address or exclude an alternative effect of GSDMD on neutrophils. In this context, it is worth mentioning that the transfer of WT neutrophils to Gsdmd−/− mice restored NET production and increased organ injury markers, reversing the protection associated with Gsdmd gene deletion. Notably, the transfer of WT monocytes to Gsdmd−/− mice slowly increased the production of cytokines, such as IL-6 and IL-1β, but the levels of organ injury markers were similar to those of the controls. Although studies have suggested that IL-1β plays a role in the physiopathology of sepsis,48-51 2 phase 3 clinical trials failed to demonstrate that the anti–IL-1β receptor antibody exhibits beneficial effects on septic patients.50,51 These results reinforce our data demonstrating that GSDMD-mediated NETs released by neutrophils are relevant to the deleterious events observed in sepsis, such as systemic inflammation and organ dysfunction.
To investigate the mechanism of GSDMD activation in neutrophils during sepsis, we explored the canonical (AIM2, NLRC4, caspase-1) and noncanonical (caspase-5/11) pathways of inflammasome activation.52,53 We observed that Aim2, Nlrc4, and caspase-1 knockout mice were not protected against CLP-induced sepsis. However, caspase-11 knockout mice were resistant to bacterial sepsis, despite expressing TLR4, a classic receptor that senses LPS from gram-negative bacteria.54 Consistently, previous studies have demonstrated that cytoplasmic caspase-11 is directly activated by LPS or gram-negative bacteria in a TLR-4–independent manner.54-56 Furthermore, the protective effect of caspase-11 deletion was associated with the abrogation of GSDMD cleavage in neutrophils and a reduction in NET release, preventing organ injury and death. Our data support the model in which GSDMD activity is downstream of initial caspase-11 activation. That caspase-11 deletion did not totally reduce GSDMD activation suggests that other putative components are required for GSDMD activation and neutrophil NETosis. In this context, Sollberg et al45 demonstrated that GSDMD is also cleaved by NE to induce NETosis. Kambara et al57 also showed that NE processes GSDMD into an active fragment. Consistent with these findings, we observed that NE is involved in GSDMD activation and NET formation after cytosolic LPS stimulation.
A recent study showed that disulfiram, a drug currently approved by the US Food and Drug Administration to treat alcohol dependence because of its inhibitory effect on ALDH37 is also a potent inhibitor of GSDMD at therapeutic doses. Disulfiram binds covalently to GSDMD, preventing pore formation in the cell membranes.12 In the present study, we demonstrated for the first time that disulfiram-mediated GSDMD inhibition in neutrophils efficiently abrogated NETosis and that treatment of septic mice with disulfiram inhibited systemic inflammation and vital organ dysfunction and improved survival.
The clinical relevance of our finding is supported by the observation that neutrophils from septic patients express more Gsdmd and produce high levels of NETs. Notably, we identified that GSDMD was associated with the structure of NETs. Furthermore, the treatment of neutrophils from patients with sepsis disulfiram prevented NET formation, suggesting that GSDMD contributes to NET formation in humans, similar to that observed in mice. Although there is controversy in the literature regarding whether the experimental sepsis models mimic the immunopathology events observed in clinical sepsis,58 the CLP model reproduces systemic inflammation, multiorgan damage, and cardiovascular collapse1,8,27,59 events investigated in the present study.
Considering that GSDMD is the effector of NETosis, promising inhibitors of GSDMD could have a pharmacologic impact on the treatment of other diseases, in addition to sepsis, in which NETs participate in immunopathology. In this context, our group and others have demonstrated that patients with COVID-19 present increased amounts of circulating and infiltrated neutrophils in lung tissue that release many NETs, which are associated with organ damage and a worse prognosis.60,61 Therefore, interventions to inhibit GSDMD would likely be effective at preventing NET-based organ injury associated with COVID-19.
Collectively, our study demonstrated that the pore-forming protein GSDMD is active in neutrophils from septic mice and humans and plays a crucial role in NET release and organ dysfunction during sepsis. Importantly, we also propose that GSDMD inhibitors, such as disulfiram, have potential clinical applications to improve sepsis therapeutic outcomes.
Acknowledgments
The authors thank Juliana Abmansur, Marcerlla Grando, Marco Antônio, Ana Kátia dos Santos, Ieda Regina, Sergio Rosa, and Diva A. M. Souza for technical assistance.
This work was supported by grants from the São Paulo Research Foundation (FAPESP) under grant agreement no. 2013/08216-2 (Center for Research in Inflammatory Diseases) and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Authorship
Contribution: C.M.S.S., C.W.S.W., F.P.V., D.S.Z., and F.Q.C. conceived and designed the study; C.M.S.S., C.W.S.W., D.C.N., A.V.G., F.P.V., D.F.C., V.F.B., F.S., T.V.M., V.S.B., K.P.S., J.E.T.-K., A.L.J.S., V.S.M., L.E.A.D., S.S.B., P.B.D., D.S.Z., M.C.B., A..E.R.O., and F.A. acquired the data (eg, provided animals, acquired and managed patients, provided facilities; C.M.S.S., C.W.S.W., H.I.N., T.M.C., J.C.A.-F., A.T.F., D.S.Z., and F.Q.C. analyzed and interpreted the data (statistical analysis, biostatistics, computational analysis; C.M.S.S., C.W.S.W., H.I.N., T.M.C., J.C.A.-F., D.S.Z., and F.Q.C. wrote, reviewed, and/or revised the manuscript; and D.S.Z. and F.Q.C. supervised the study.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Fernando Q. Cunha, Av Bandeirantes 3900, Monte Alegre 14049–900, Ribeirão Preto, SP, Brazil; e-mail: fdqcunha@fmrp.usp.br; and Camila Meirelles S. Silva, Av Bandeirantes 3900, Monte Alegre 14049–900, Ribeirão Preto, SP, Brazil; e-mail: camilameirelles.s@usp.br.
For original data, please contact the corresponding authors.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal