

Key Points
DYRK1a is a BAFF-responsive kinase that mediates noncanonical NF-κB activation, B-cell survival, and B-cell autoimmunity.
The DYRK1a noncanonical NF-κB signaling axis is important for B-ALL cell survival.

Abstract
B-cell–activating factor (BAFF) mediates B-cell survival and, when deregulated, contributes to autoimmune diseases and B-cell malignancies. The mechanism connecting BAFF receptor (BAFFR) signal to downstream pathways and pathophysiological functions is not well understood. Here we identified DYRK1a as a kinase that responds to BAFF stimulation and mediates BAFF-induced B-cell survival. B-cell–specific DYRK1a deficiency causes peripheral B-cell reduction and ameliorates autoimmunity in a mouse model of lupus. An unbiased screen identified DYRK1a as a protein that interacts with TRAF3, a ubiquitin ligase component mediating degradation of the noncanonical nuclear factor (NF)-κB–inducing kinase (NIK). DYRK1a phosphorylates TRAF3 at serine-29 to interfere with its function in mediating NIK degradation, thereby facilitating BAFF-induced NIK accumulation and noncanonical NF-κB activation. Interestingly, B-cell acute lymphoblastic leukemia (B-ALL) cells express high levels of BAFFR and respond to BAFF for noncanonical NF-κB activation and survival in a DYRK1a-dependent manner. Furthermore, DYRK1a promotes a mouse model of B-ALL through activation of the noncanonical NF-κB pathway. These results establish DYRK1a as a critical BAFFR signaling mediator and provide novel insight into B-ALL pathogenesis.
Introduction
B cells play a central role in mediating humoral immune responses and, when deregulated, contribute to the development of autoimmune diseases and B-cell malignancies.1-3 Early stages of B-cell development occur in the bone marrow, where B-cell progenitor cells progress through pro-B, pre-B, and immature B-cell stages defined by ordered rearrangement and expression of B-cell receptor (BCR) genes.4 Immature B cells migrate to the spleen as transitional 1 (T1) B cells, which undergo a process of maturation to become T2 immature cells and then mature B cells under the regulation of signals from the BCR and B-cell activating factor (BAFF) receptor (BAFFR).5 BAFFR is particularly important for the survival of T2 and mature B cells.6 Defect in BAFF expression or BAFFR signaling is associated with humoral immune deficiencies, whereas aberrant BAFF production contributes to the pathogenesis of autoimmune diseases.7
A major signaling event stimulated by the BAFFR is activation of the noncanonical nuclear factor (NF)-κB (ncNF-κB) pathway, which is critical for peripheral B-cell development and survival.8 This pathway is based on proteasomal processing of the NF-κB2 precursor protein p100, which generates mature NF-κB2 and p52 and causes nuclear translocation of p100-sequestered NF-κB members, p52 and RelB.9,10 A central component of the ncNF-κB pathway is NF-κB–inducing kinase (NIK), encoded by the gene Map3k14, which along with its downstream kinase, IKKα, induces phosphorylation-dependent processing of p100.9,11 The mechanism of ncNF-κB signaling centers on the regulation of NIK degradation. Under steady-state conditions, NIK is targeted for ubiquitin-dependent degradation by an E3 ubiquitin ligase complex, composed of cIAP, TRAF2, and TRAF3 (hereafter called cIAP-TRAF complex), which serves as a mechanism to prevent signal-independent ncNF-κB activation.10,12-15 In response to signals from a subset of tumor necrosis factor receptor superfamily members, which in B cells include BAFFR and CD40, NIK is liberated from this E3 ligase complex, and the accumulation of stabilized NIK triggers ncNF-κB activation.10 Genetic deficiencies in ncNF-κB signaling components can cause immunodeficiencies, whereas deregulated activation of this pathway is associated with autoimmune diseases and B-cell malignancies.10,16-18 However, to date, the molecular mechanism underlying signal-induced NIK stabilization is still poorly defined.
As an approach to elucidate the mechanism underlying the regulation of cIAP-TRAF E3 ligase complex, we performed an unbiased screening to identify novel factors physically associating with TRAF3. We identified a novel TRAF3-binding protein, dual-specificity tyrosine phosphorylation-regulated kinase 1a (DYRK1a). DYRK1a gene locus amplification has been associated with human B-cell acute lymphoblastic leukemia (B-ALL), and DYRK1a is important for leukemogenesis in a mouse model of B-ALL.19,20 Notably, although B-ALL cells are of pre–B-cell origin, they are characterized by a high level of BAFFR expression.21 We show in the present study that DYRK1a is a kinase that responds to BAFFR and CD40 stimulation and mediates ncNF-κB activation and peripheral B-cell survival. Our data suggest that DYRK1a promotes NIK stabilization by phosphorylating TRAF3. These findings establish DYRK1a as a novel mediator of ncNF-κB activation and provide new insight into peripheral B-cell development and B-ALL pathogenesis.
Methods
BioID screening of TRAF3-interacting proteins
M12 cells were transduced with a retroviral vector encoding biotin identification (BioID) TRAF3 fusion protein with an N-terminal Myc epitope tag (pCLXSN(GFP)-myc-BioID-TRAF3) or the control pCLXSN-myc-BioID vector. The cells were cultured in biotin-supplemented medium for 24 h and then subjected to BioID screening, as described previously.22 TRAF3-interacting proteins were identified using mass spectrometry at the Mass Spectrometry Proteomics Core of Baylor College of Medicine (Houston, TX).
B-cell purification and in vitro analysis
B cells were purified from the splenocytes by anti–B220-conjugated magnetic beads (Miltenyl Biotec). Purified B cells in replicate wells of 96-well plates (2 × 105 cells per well) were stimulated at 37°C with CD40L (500 ng/mL) or BAFF (200 ng/mL). After stimulation for 40 h, B cells were pulse-labeled for 6 h with [3H] thymidine and then collected for thymidine-incorporation assays. B-cell survival was analyzed by SYTOX Blue dead cell stain (Invitrogen) after 48-h culture with indicated condition. APC-annexin V and propidium iodide (BD Pharmingen) were used to quantify the apoptotic cell population.
Flow cytometry
Cell suspensions from various organs or cell lines were stained as indicated and acquired using an LSRFortessa flow cytometer (BD Biosciences), as described previously.23 The data were analyzed using FlowJo software. Gating strategy is shown in supplemental Figure 1 (available on the Blood Web site).
BM12 model of lupus-like autoimmune disease
The BM12 model of lupus-like autoimmune disease was induced and analyzed as described.22
Statistical analysis
Statistical analysis was performed using GraphPad Prism software. Significant changes between 2 groups were analyzed with an unpaired, 2-tailed Student t test. Two-way analysis of variance (ANOVA) with Bonferroni multiple-comparison test were used for in vitro cell survival assay. Kaplan-Meier analyses were performed, and the log-rank Mantel-Cox test was used to determine statistical difference between the survival curves of two groups. Values of P < .05 were considered significant, and the level of significance was indicated as *P < .05, **P < .01, and ***P < .001. All data are presented as mean ± standard deviation (SD).
Results
DYRK1a is a TRAF3-binding protein
To characterize novel regulators and functions of the ncNF-κB signaling pathway, we screened for TRAF3-binding proteins by proximity-dependent BioID, a technique that allows identification of signaling partners–based in vivo protein association.24 We constructed retroviral vectors encoding either a Myc-BioID control or Myc-BioID–TRAF3 fusion protein and stably infected a B-cell line, M12 (supplemental Figure 2A-B). After biotin treatment of the cells, we performed streptavidin bead pull-down to isolate the biotinylated proteins (supplemental Figure 2C-D). Mass spectrometry analysis identified several known TRAF3-binding proteins, such as TBK1, CYLD, TRAF5, as well as novel TRAF3-interactors, including DYRK1a (supplemental Table 1).
Co-immunoprecipitation (CoIP) assays confirmed the strong interaction between TRAF3 and DYRK1a (Figure 1B,D) and revealed the essential role of the C-terminal TRAF domain (TRAF-C) and a polyhistidine motif of DYRK1a for this molecular interaction (Figure 1A-E). The interaction between TRAF3 and DYRK1a was also readily detected under endogenous conditions in B cells, and this interaction was constitutive and not further enhanced upon BAFF stimulation (Figure 1F). These results identify DYRK1a as a novel factor physically interacting with TRAF3.
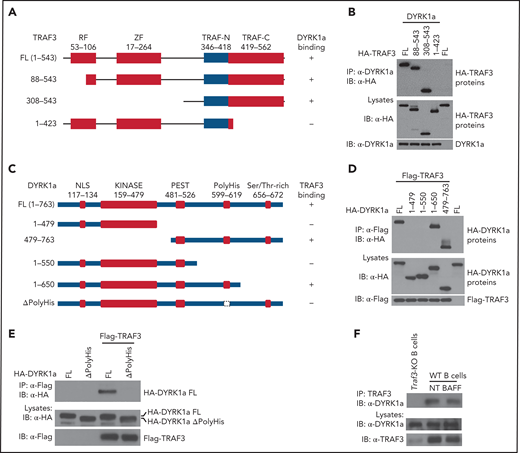
Domain-specific interaction between DYRK1a and TRAF3. (A) Schematic summary of TRAF3 and its truncation mutants, depicting the ring finger (RF), zinc finger (ZF), and TRAF (TRAF-N and TRAF-C) domains and indicating their DYRK1a-binding function based on the CoIP results from panel B. (B) CoIP analysis of DYRK1a interaction with TRAF3 mutants using whole-cell lysates of HEK293 cells transfected with the indicated expression vectors (A). Cell lysates were also subjected to direct immunoblotting to monitor expression of TRAF3 mutants and DYRK1a (B). (C) Schematic representation depicting the domains of DYRK1a and its mutants and their ability to bind TRAF3 (based on CoIP results of D and E). (D-E) CoIP analysis of TRAF3 interaction with DYRK1a mutants using whole-cell lysates of HEK293 cells transfected with the indicated expression vectors (C). Cell lysates were also subjected to direct immunoblotting to monitor expression of DYRK1a mutants and TRAF3 (D,E). (F) CoIP assays to analyze the interaction of endogenous TRAF3 and DYRK1a in nontreated (NT) or BAFF-stimulated WT B cells. Nontreated TRAF3-BKO B cells were used as negative control.
Domain-specific interaction between DYRK1a and TRAF3. (A) Schematic summary of TRAF3 and its truncation mutants, depicting the ring finger (RF), zinc finger (ZF), and TRAF (TRAF-N and TRAF-C) domains and indicating their DYRK1a-binding function based on the CoIP results from panel B. (B) CoIP analysis of DYRK1a interaction with TRAF3 mutants using whole-cell lysates of HEK293 cells transfected with the indicated expression vectors (A). Cell lysates were also subjected to direct immunoblotting to monitor expression of TRAF3 mutants and DYRK1a (B). (C) Schematic representation depicting the domains of DYRK1a and its mutants and their ability to bind TRAF3 (based on CoIP results of D and E). (D-E) CoIP analysis of TRAF3 interaction with DYRK1a mutants using whole-cell lysates of HEK293 cells transfected with the indicated expression vectors (C). Cell lysates were also subjected to direct immunoblotting to monitor expression of DYRK1a mutants and TRAF3 (D,E). (F) CoIP assays to analyze the interaction of endogenous TRAF3 and DYRK1a in nontreated (NT) or BAFF-stimulated WT B cells. Nontreated TRAF3-BKO B cells were used as negative control.
DYRK1a is required for maintenance of peripheral B cells
TRAF3 controls B-cell survival and maturation through regulating ncNF-κB activation.25-27 We therefore examined the role of DYRK1a in regulating B-cell development and maturation by generating DYRK1a B-cell–conditional knockout (Dyrk1aBKO) mice. We crossed the Dyrk1a-flox mice with Cd19-Cre or Mb1-Cre mice (supplemental Figure 3); the Cd19-Cre mice are known to express the Cre recombinase predominantly in peripheral B cells, whereas the Mb1-Cre mice strongly express the Cre recombinase in early stages of bone marrow B cells.28 In line with a role of DYRK1a in pre–B-cell development,29 deletion of DYRK1a using the Mb1-Cre reduced the number of bone marrow B cells from the pre-B stage (supplemental Figure 4A-B). Interestingly, a more striking defect was seen with the mature B cells (supplemental Fig. 4A-B). Because mature B cells are generated in the spleen, we examined the role of DYRK1a in regulating peripheral B cells by using the Dyrk1aBKO mice generated with Cd19-Cre. Consistent with the poor expression of Cd19-Cre in early stages of bone marrow B cells, deletion of DYRK1a using this Cre did not significantly reduce the number of pro-B, pre-B, or immature B cells but led to a reduction in the mature B-cell population (supplemental Figure 4C-D). We used this BKO model for the following studies except as otherwise indicated. Compared with wild-type (WT) control mice, the Dyrk1aBKO mice had a decreased frequency and absolute number of B cells in peripheral lymphoid organs (Figure 2A). The DYRK1a deficiency predominantly reduced the number of mature B cells, although it also moderately reduced the number of immature B cells (Figure 2B).
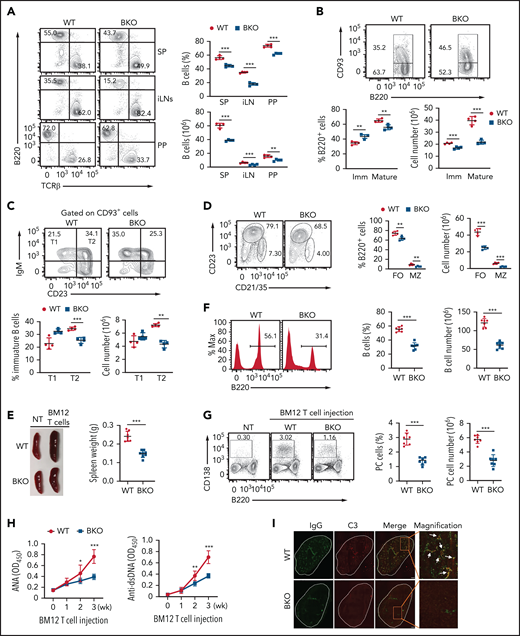
DYRK1a is essential for peripheral B-cell maintenance and B-cell–mediated autoimmunity. (A) Flow cytometric analyses of B220+ B cells and T-cell receptor (TCR) β+ T cells in the spleen (SP), inguinal lymph nodes (iLN), and Peyer’s patches (PP) of WT or Dyrk1aBKO mice (6-8 weeks old). (B-D) Flow cytometric analyses of immature (Imm; B220+CD93+) and mature (B220+CD93−) B cells (B), T1 (CD93+IgM+CD23–) and T2 (CD93+IgM+CD23+) immature B cells (C), and follicular (FO, B220+CD21intCD23+) and marginal zone (MZ; B220+CD21hiCD23−) B cells (D) in the spleen of WT and Dyrk1aBKO mice (6-8 weeks old). (E) A representative spleen image (left) and a summary graph of spleen weight (right, each circle represents a mouse) of WT and Dyrk1aBKO mice that were either not treated (NT) or injected with CD4 T cells from BM12 mice for 3 weeks. (F-G) Flow cytometric analysis of B220+ B cells (F) and B220−CD138+ plasma cells (G) in WT and Dyrk1aBKO mice that were injected with BM12 CD4 T cells for 3 weeks. Data are presented as a representative plot (left) and summary graph (right) based on multiple recipient mice. (H) ELISA of autoantibodies reacting against double-stranded DNA (anti-dsDNA) and nuclear antigen (ANA) in serum from WT and Dyrk1aBKO mice injected with BM12 CD4 T cells for the indicated time periods. (I) Confocal microscopic analyses of glomerular IgG and C3 deposition in kidney sections from WT and Dyrk1aBKO mice injected with BM12 CD4 T cells for 3 weeks. Scale bar, 1 mm. The arrows depict colocalization of IgG and C3. Data are representative of 3 independent experiments. Summary graphs are presented as mean ± SD, and P values were determined by an unpaired, 2-tailed Student t test (A-B,H). *P < .05; **P < .01; ***P < .001.
DYRK1a is essential for peripheral B-cell maintenance and B-cell–mediated autoimmunity. (A) Flow cytometric analyses of B220+ B cells and T-cell receptor (TCR) β+ T cells in the spleen (SP), inguinal lymph nodes (iLN), and Peyer’s patches (PP) of WT or Dyrk1aBKO mice (6-8 weeks old). (B-D) Flow cytometric analyses of immature (Imm; B220+CD93+) and mature (B220+CD93−) B cells (B), T1 (CD93+IgM+CD23–) and T2 (CD93+IgM+CD23+) immature B cells (C), and follicular (FO, B220+CD21intCD23+) and marginal zone (MZ; B220+CD21hiCD23−) B cells (D) in the spleen of WT and Dyrk1aBKO mice (6-8 weeks old). (E) A representative spleen image (left) and a summary graph of spleen weight (right, each circle represents a mouse) of WT and Dyrk1aBKO mice that were either not treated (NT) or injected with CD4 T cells from BM12 mice for 3 weeks. (F-G) Flow cytometric analysis of B220+ B cells (F) and B220−CD138+ plasma cells (G) in WT and Dyrk1aBKO mice that were injected with BM12 CD4 T cells for 3 weeks. Data are presented as a representative plot (left) and summary graph (right) based on multiple recipient mice. (H) ELISA of autoantibodies reacting against double-stranded DNA (anti-dsDNA) and nuclear antigen (ANA) in serum from WT and Dyrk1aBKO mice injected with BM12 CD4 T cells for the indicated time periods. (I) Confocal microscopic analyses of glomerular IgG and C3 deposition in kidney sections from WT and Dyrk1aBKO mice injected with BM12 CD4 T cells for 3 weeks. Scale bar, 1 mm. The arrows depict colocalization of IgG and C3. Data are representative of 3 independent experiments. Summary graphs are presented as mean ± SD, and P values were determined by an unpaired, 2-tailed Student t test (A-B,H). *P < .05; **P < .01; ***P < .001.
Immature B cells in the spleen include transitional (T) 1 and T2 populations.6 The B-cell–specific DYRK1a deficiency caused a reduction in the T2, but not T1, population (Figure 2C), suggesting the requirement of DYRK1a for the maintenance of T2, but not T1, stage of immature B cells. Splenic mature B cells include follicular and marginal zone B-cell populations.6 Both the follicular and the marginal zone B cells were reduced in the Dyrk1aBKO mice (Figure 2D). These results demonstrate a crucial role of DYRK1a in the generation or maintenance of mature B cells and T2 immature B cells. Of note, these phenotypes of the Dyrk1aBKO mice were reminiscent of mutant mice defective in ncNF-κB signaling.6
DYRK1a is required for B-cell–mediated autoimmunity
B cells are a major mediator of systemic autoimmune diseases, including systemic lupus erythematosus (SLE), and manipulation of B cells has been explored as a therapeutic approach. Because of the requirement of DYRK1a in peripheral B-cell maintenance, we examined the role of DYRK1a in autoimmunity using a mouse model of SLE.30 This model involves adoptive transfer of T cells from a C57BL/6 variant mouse strain, BM12, harboring a 3-amino acid substitution in the MHCII molecule to the WT or Dyrk1aBKO mice (in C57BL/6 background). The BM12 donor T cells become activated in the recipient via alloreactivity and then activate self-reactive recipient B cells to cause SLE-like symptoms. Upon BM12 T-cell transfer, the WT mice developed SLE-like symptoms, including enlargement of the spleen, B-cell hyperplasia associated with increased plasma cells, autoantibody accumulation, and deposition of immunoglobulin (Ig) G/complement immune complexes to the kidney glomeruli (Figure 2E-I). Importantly, these phenotypes were ameliorated in the Dyrk1aBKO recipient mice (Figure 2E-I). In a control experiment, transplantation of the WT and Dyrk1aBKO mice with C57BL/6 T cells did not cause plasma cell increase or autoantibody production (supplemental Fig. 5). These results suggest that DYRK1a is critical for the induction of SLE-like autoimmune symptoms.
DYRK1a mediates ncNF-κB activation and B-cell survival
The results described above raised the question of whether DYRK1a might regulate B-cell survival or expansion induced by ncNF-κB inducers. As expected, wild-type B cells underwent massive cell death when cultured ex vivo, which was inhibited by the ncNF-κB inducers, BAFF and CD40L (Figure 3A). Importantly, DYRK1a deficiency impaired BAFF- and CD40L-induced B-cell survival and proliferation (Figure 3B-C).
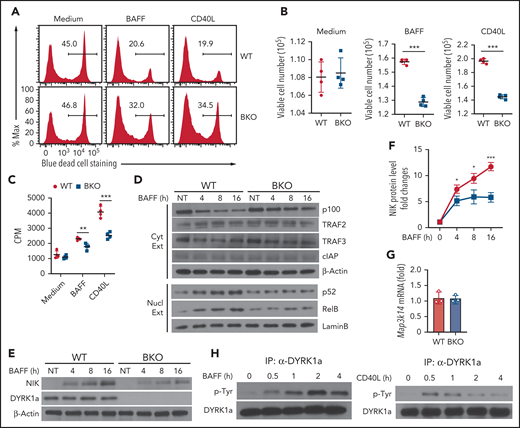
DYRK1a mediates noncanonical NF-κB activation and B-cell survival. (A-C) B cells purified from the spleen of WT and Dyrk1aBKO mice were cultured for 48 h in 96-well plate (2 × 105 cells per well), in the presence of medium control, BAFF or CD40L. The cells were subjected to flow cytometry to quantity dead and viable cell populations, presented as a representative plot (A) and summary graphs based on multiple mice (B), or proliferation assays based on 3H-thymidine incorporation (C). (D) Immunoblot analysis of the indicated proteins using cytoplasmic (Cyt) or nuclear (Nucl) extracts of BAFF-stimulated WT and Dyrk1aBKO splenic B cells. (E-F) Immunoblot analysis of the indicated proteins in whole-cell extracts of BAFF-stimulated WT and Dyrk1aBKO splenic B cells. Data are presented as a representative blot (E) and a summary graph of densitometric quantification of the NIK protein bands based on 3 independent experiments (F). NIK level was quantified as ratio to β-Actin and presented as fold to the nontreated (NT) lane (set as 1). (G) qRT-PCR analysis of Map3k14 mRNA in WT or Dyrk1aBKO splenic B cells stimulated with BAFF for 16 h. (H) DYRK1a was immunoprecipitated from BAFF- or CD40L-stimulated WT B cells and subjected to immunoblot assays using anti-phosphorylated tyrosine (p-Tyr) or DYRK1a. Summary graphs are presented as mean ± SD, and P values were determined by an unpaired, 2-tailed Student t test (B-C,F). *P < .05; **P < .01; ***P < .001.
DYRK1a mediates noncanonical NF-κB activation and B-cell survival. (A-C) B cells purified from the spleen of WT and Dyrk1aBKO mice were cultured for 48 h in 96-well plate (2 × 105 cells per well), in the presence of medium control, BAFF or CD40L. The cells were subjected to flow cytometry to quantity dead and viable cell populations, presented as a representative plot (A) and summary graphs based on multiple mice (B), or proliferation assays based on 3H-thymidine incorporation (C). (D) Immunoblot analysis of the indicated proteins using cytoplasmic (Cyt) or nuclear (Nucl) extracts of BAFF-stimulated WT and Dyrk1aBKO splenic B cells. (E-F) Immunoblot analysis of the indicated proteins in whole-cell extracts of BAFF-stimulated WT and Dyrk1aBKO splenic B cells. Data are presented as a representative blot (E) and a summary graph of densitometric quantification of the NIK protein bands based on 3 independent experiments (F). NIK level was quantified as ratio to β-Actin and presented as fold to the nontreated (NT) lane (set as 1). (G) qRT-PCR analysis of Map3k14 mRNA in WT or Dyrk1aBKO splenic B cells stimulated with BAFF for 16 h. (H) DYRK1a was immunoprecipitated from BAFF- or CD40L-stimulated WT B cells and subjected to immunoblot assays using anti-phosphorylated tyrosine (p-Tyr) or DYRK1a. Summary graphs are presented as mean ± SD, and P values were determined by an unpaired, 2-tailed Student t test (B-C,F). *P < .05; **P < .01; ***P < .001.
Remarkably, the DYRK1a deficiency attenuated BAFF-induced ncNF-κB activation, as revealed by the impaired loss of cytoplasmic p100 and induction of nuclear p52 and RelB (Figure 3D). A central step in ncNF-κB signaling is stabilization of NIK, which is normally caused by degradation of its inhibitory protein TRAF3.31 Interestingly, the DYRK1a deficiency did not inhibit TRAF3 degradation. Nevertheless, the DYRK1a deficiency impaired induction of NIK protein accumulation (Figure 3E-F) without inhibiting Map3k14 mRNA expression (Figure 3G). These results, along with the DYRK1a-TRAF3 interaction, suggested a role for DYRK1a in regulating ncNF-κB signaling.
Because DYRK1a regulates signal-induced, but not steady-state, ncNF-κB activation and B-cell survival, we questioned whether this kinase responds to the ncNF-κB signals. In this regard, DYRK1a activation involves its autophosphorylation at a conserved tyrosine residue in the activation loop.32 Importantly, while the basal level of DYRK1a phosphorylation was low in B cells, it was potently stimulated by BAFF and CD40L (Figure 3H). These results establish DYRK1a as a kinase that facilitates the induction of ncNF-κB signaling and B-cell survival by BAFFR and CD40.
Ectopic NIK expression rescues the B-cell defect of Dyrk1aBKO mice
To determine the role of the ncNF-κB pathway in DYRK1a-mediated B-cell regulation, we used the R26StopFLMap3k14 transgenic mice carrying a Map3k14 transgene under the control of a loxP-flanked STOP cassette.33 By crossing these mice with the Dyrk1aBKO mice, we generated mutant mice with B-cell–specific NIK overexpression and DYRK1a deletion (Dyrk1aBKOMap3k14Btg). Remarkably, while the Dyrk1aBKO mice had reduced frequency and number of splenic B-cell populations, this phenotype was reversed in the Dyrk1aBKOMap3k14Btg mice (Figure 4A-B). Consistently, NIK expression rescued the activation of ncNF-κB in DYRK1a-deficient B cells and restored the survival ability of these mutant B cells (Figure 4C-E). Compared with the WT mice, the Map3k14Btg mice had increased frequency and number of B cells (supplemental Figure 6A-B), which was in line with a previous study.33 Importantly, DYRK1a deficiency in the Map3k14Btg mice (Dyrk1aBKOMap3k14Btg) did not cause a reduction in B-cell populations (supplemental Figure 6A-B), suggesting that ectopic NIK expression rescues the B-cell defect of DYRK1a deficiency. Compared with WT control B cells, the Map3k14Btg B cells also displayed increased survival, which was only moderately reduced upon DYRK1a deletion (Map3k14BtgDyrk1aBKO) (supplemental Figure 6C-D). Consistently, NIK ectopic expression led to strong ncNF-κB activation (nuclear p52 expression) in both the Map3k14Btg and Map3k14BtgDyrk1aBKO B cells (supplemental Figure 6E), suggesting that NIK functions downstream of DYRK1a. Thus, DYRK1a mediates peripheral B-cell survival and maturation via facilitating activation of the ncNF-κB pathway.
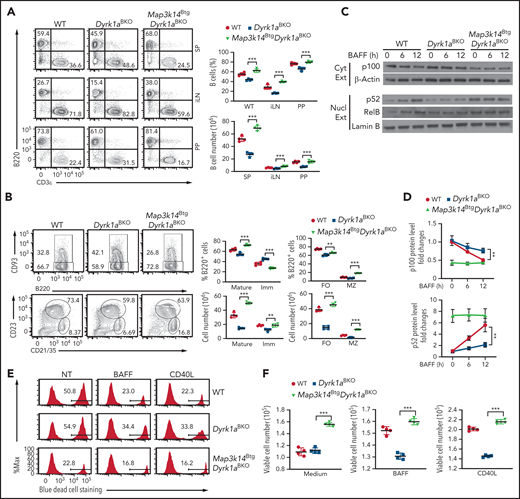
Ectopic expression of NIK rescues the defect of Dyrk1aBKO mice in peripheral B-cell development and survival. (A) Flow cytometric analyses of B220+ B cells and TCRβ+ T cells (A), immature (Imm; B220+CD93+) and mature (B220+CD93−) B cells as well as follicular (FO; B220+CD21intCD23+) and marginal zone (MZ; B220+CD21hiCD23−) B cells (B) in the spleen (SP), inguinal lymph nodes (iLN), and Peyer’s patches (PP) of wild-type, Dyrk1aBKO, and Map3k14BtgDyrk1aBKO mice (6-8 weeks old). (C-D) Immunoblot analysis of the indicated proteins in the cytoplasmic (Cyt Ext) or nuclear (Nucl Ext) extracts of wild-type, Dyrk1aBKO, and Map3k14BtgDyrk1aBKO splenic B cells stimulated with BAFF. Data are presented as a representative blot (C) and summary graphs of densitometrically quantified cytoplasmic p100 (ratio to β-Actin) and nuclear p52 (ratio to Lamin B) protein bands, presented as fold to 0 time point value (set to 1) (D). (E-F) Purified wild-type, Dyrk1aBKO, and Map3k14BtgDyrk1aBKO splenic B cells were cultured for 48 h in 96-well plates (2 × 105 cells per well) in the presence of medium control, BAFF, or CD40L and subjected to flow cytometry to quantify dead and viable cells. Data are presented as representative plots (E) and summary graphs (F). Data are representative of 3 independent experiments, and summary graphs were presented as mean ± SD with P values determined by an unpaired, 2-tailed Student t test (A-B,D,F). *P < .05; **P < .01; ***P < .001.
Ectopic expression of NIK rescues the defect of Dyrk1aBKO mice in peripheral B-cell development and survival. (A) Flow cytometric analyses of B220+ B cells and TCRβ+ T cells (A), immature (Imm; B220+CD93+) and mature (B220+CD93−) B cells as well as follicular (FO; B220+CD21intCD23+) and marginal zone (MZ; B220+CD21hiCD23−) B cells (B) in the spleen (SP), inguinal lymph nodes (iLN), and Peyer’s patches (PP) of wild-type, Dyrk1aBKO, and Map3k14BtgDyrk1aBKO mice (6-8 weeks old). (C-D) Immunoblot analysis of the indicated proteins in the cytoplasmic (Cyt Ext) or nuclear (Nucl Ext) extracts of wild-type, Dyrk1aBKO, and Map3k14BtgDyrk1aBKO splenic B cells stimulated with BAFF. Data are presented as a representative blot (C) and summary graphs of densitometrically quantified cytoplasmic p100 (ratio to β-Actin) and nuclear p52 (ratio to Lamin B) protein bands, presented as fold to 0 time point value (set to 1) (D). (E-F) Purified wild-type, Dyrk1aBKO, and Map3k14BtgDyrk1aBKO splenic B cells were cultured for 48 h in 96-well plates (2 × 105 cells per well) in the presence of medium control, BAFF, or CD40L and subjected to flow cytometry to quantify dead and viable cells. Data are presented as representative plots (E) and summary graphs (F). Data are representative of 3 independent experiments, and summary graphs were presented as mean ± SD with P values determined by an unpaired, 2-tailed Student t test (A-B,D,F). *P < .05; **P < .01; ***P < .001.
DYRK1a regulates TRAF3 function via phosphorylation
Because DYRK1a is a kinase interacting with TRAF3, we examined whether DYRK1a might phosphorylate TRAF3 by mass spectrometric analysis of TRAF3 co-expressed in 293 with WT DYRK1a or a kinase-dead DYRK1a mutant, K188R (supplemental Figure 7A). We identified 3 major phosphorylation sites, S9, T29, and S75, in TRAF3, among which T29 was specifically induced by the WT DYRK1a (supplemental Figure 7B-C; supplemental Table 2). T29 is located in the N-terminal region of human TRAF3, and this site is conserved as either a T or S across vertebrate species (supplemental Figure 7D). T29 and S29 were also identified as phosphorylation sites of human and mouse TRAF3, respectively, in public phosphorylation database (supplemental Figure 7E). In vitro kinase assays confirmed that purified DYRK1A could phosphorylate the N-terminal region (amino acid 1-165) of both human and mouse TRAF3 (Figure 5A-B). Furthermore, mutation of T29 in human TRAF3 (1-165) and S29 in mouse TRAF3 (1-165) abolished TRAF3 phosphorylation by DYRK1a (Figure 5A-B). Using the in vitro kinase assay, we further demonstrated that the catalytic activity of endogenous DYRK1a was stimulated in B cells by BAFF and CD40L (Figure 5C).
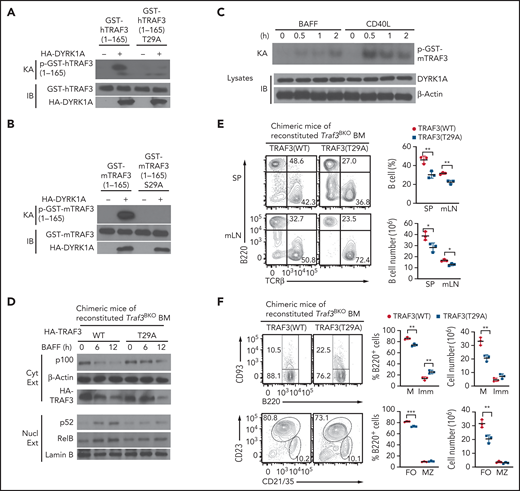
DYRK1a phosphorylates TRAF3. (A-B) In vitro kinase assays using HA-DYRK1a isolated by IP from transfected 293 cells and the indicated human (A) or murine (B) GST-TRAF3 recombinant proteins as substrates. The kinase assay membrane was subjected to IB to detect HA-DYRK1a and the GST-TRAF3 substrates. (C) In vitro kinase assays using endogenous DYRK1a isolated by IP from BAFF- or CD40L-stimulated WT B cells and murine GST-TRAF3 substrate. (D) Chimeric mice were generated by adoptively transferring Rag1−/− mice with Traf3BKO bone marrow cells transduced with expression vectors for WT TRAF3 or T29A mutant. Immunoblot was performed to analyze the indicated proteins using cytoplasmic or nuclear extracts of BAFF-stimulated splenic B cells isolated from chimeric mice of Traf3BKO bone marrow reconstituted with TRAF3 WT or T29A. (E) Flow cytometric analysis of the B220+ B cells and TCRβ+ T cells in the spleen (SP) or mesenteric lymph nodes (mLN) of the indicated chimeric mice described in panel D. (F) Flow cytometric analysis of the mature (M; B220+CD93−), immature (Imm; B220+CD93+), follicular (FO; B220+CD21intCD23+), and marginal zone (MZ; B220+CD21hiCD23−) B cells in the spleen of the indicated chimeric mice described in D. Data are presented as a representative plot, and summary graphs are mean ± SD values based on multiple mice, and P values are determined by an unpaired, 2-tailed Student t test. *P < .05; **P < .01; ***P < .001.
DYRK1a phosphorylates TRAF3. (A-B) In vitro kinase assays using HA-DYRK1a isolated by IP from transfected 293 cells and the indicated human (A) or murine (B) GST-TRAF3 recombinant proteins as substrates. The kinase assay membrane was subjected to IB to detect HA-DYRK1a and the GST-TRAF3 substrates. (C) In vitro kinase assays using endogenous DYRK1a isolated by IP from BAFF- or CD40L-stimulated WT B cells and murine GST-TRAF3 substrate. (D) Chimeric mice were generated by adoptively transferring Rag1−/− mice with Traf3BKO bone marrow cells transduced with expression vectors for WT TRAF3 or T29A mutant. Immunoblot was performed to analyze the indicated proteins using cytoplasmic or nuclear extracts of BAFF-stimulated splenic B cells isolated from chimeric mice of Traf3BKO bone marrow reconstituted with TRAF3 WT or T29A. (E) Flow cytometric analysis of the B220+ B cells and TCRβ+ T cells in the spleen (SP) or mesenteric lymph nodes (mLN) of the indicated chimeric mice described in panel D. (F) Flow cytometric analysis of the mature (M; B220+CD93−), immature (Imm; B220+CD93+), follicular (FO; B220+CD21intCD23+), and marginal zone (MZ; B220+CD21hiCD23−) B cells in the spleen of the indicated chimeric mice described in D. Data are presented as a representative plot, and summary graphs are mean ± SD values based on multiple mice, and P values are determined by an unpaired, 2-tailed Student t test. *P < .05; **P < .01; ***P < .001.
To determine the functional significance of DYRK1a-mediated TRAF3 phosphorylation, we generated bone marrow chimeric mice by adoptively transferring the lymphocyte-deficient Rag1-KO mice with TRAF3-deficient bone marrow cells reconstituted with either WT TRAF3 or TRAF3 T29A. Spleen B cells from the TRAF3 T29A chimeric mice had reduced processing of p100 and nuclear translocation of p52 and RelB compared with the B cells of WT TRAF3 chimeric mice (Figure 5D). Consistently, the TRAF3 T29A chimeric mice displayed a reduction in the percentage and absolute number of peripheral B cells, most strikingly mature follicular B cells (Figure 5E-F). Together, these results suggest that DYRK1a mediates ncNF-κB activation and peripheral B-cell development via phosphorylation of TRAF3.
The DYRK1a-NIK signaling axis regulates B-ALL cell survival
DYRK1a has been implicated in human leukemia and recently shown to be important for a mouse model of B-ALL.19,20 Our analysis of a human pediatric ALL database revealed DYRK1a amplification or mRNA overexpression in 10.83% of total cases (203 samples from 154 patients) (supplemental Figure 8). Notably, a hallmark of B-ALL cells is high-level expression of BAFFR despite their pre–B-cell origin.21 Because BAFFR is normally expressed in mature and T2 transitional B cells but not pre-B cells,6 its expression in malignant pre-B cells suggests the involvement of BAFFR signaling in B-ALL development. In support of the previous study, our flow cytometric assays revealed BAFFR expression in several human B-ALL cell lines, including SEMK2, TANOUE, CALL4, MTUZ5, and BALL1 (Figure 6A). Consistent with the lack of BAFFR expression in Burkitt lymphoma,34 BAFFR was not detected in the Burkitt lymphoma cell line DAUDI (Figure 6A). Using the BALL1 cell line, we demonstrated that as seen in normal B cells, DYRK1a and TRAF3 physically interacted in B-ALL cells (supplemental Figure 9). Furthermore, the BALL1 cells responded to BAFF for activation of ncNF-κB, as revealed by cytoplastic p100 degradation and nuclear p52 induction (Figure 6B). The BAFF-induced ncNF-κB activation was impaired upon DYRK1A knockdown in BALL1 cells (Figure 6B). BAFF-induced, although not steady-state, BALL1 cell survival was compromised by the DYRK1A knockdown (Figure 6C). Remarkably, ectopic NIK expression efficiently rescued the survival of DYRK1A-knockdown BALL1 cells (Figure 6C), suggesting a DYRK1a-NIK signaling axis in mediating B-ALL cell survival.
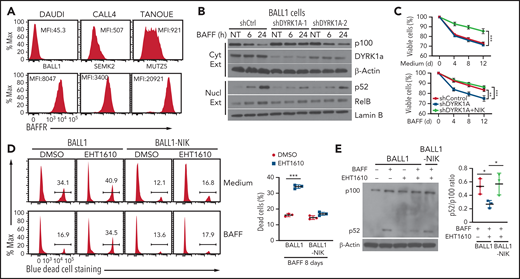
DYRK1a-NIK axis mediates B-ALL cell survival. (A) Flow cytometric analysis of BAFFR expression in human B-ALL cells. The Burkitt lymphoma cell line DAUDI was used as a negative control. (B) Immunoblot analysis of the indicated proteins in the cytoplasmic or nuclear extracts of BALL1 cells transduced with either a control nonsilencing shRNA or two different DYRK1A shRNAs and stimulated with BAFF. (C) Viable cell quantification by flow cytometry, based on SYTOX blue cell staining, of control BALL1 cells (shControl), DYRK1A-knockdown (using DYRK1A shRNA #1) BALL1 cells (shDYRK1A), or DYRK1A-knockdown BALL1 cells and transduced with the NIK expression vector (shDYRK1A + NIK), cultured in the presence of BAFF or medium control. Data are mean ± SD values combined from the mean values of 3 independent experiments. (D) Flow cytometric analysis of viable and dead cells, based on SYTOX blue cell staining, in BALL1 cells or BALL1 cells transduced with a NIK expression vector (BALL1-NIK). The cells were cultured for 8 days in the presence of BAFF or medium control along with the DYRK1a inhibitor EHT1610 (10 µM) or DMSO. The summary graph is based on 3 replicate samples of the same experiment. (E) Immunoblot analysis of p100 and its processing product p52 in BALL1 or BALL1-NIK cells cultured for 8 days in the presence (+) or absence (–) of BAFF and EHT1610. Summary graph presents p52/p100 ratio based on densitometric quantification of immunoblot protein bands in 3 independent experiments. P values are determined by an unpaired, 2-tailed Student t test (D-E) or 2-way ANOVA with Bonferroni correction (C). *P < .05; **P < .01; ***P < .001.
DYRK1a-NIK axis mediates B-ALL cell survival. (A) Flow cytometric analysis of BAFFR expression in human B-ALL cells. The Burkitt lymphoma cell line DAUDI was used as a negative control. (B) Immunoblot analysis of the indicated proteins in the cytoplasmic or nuclear extracts of BALL1 cells transduced with either a control nonsilencing shRNA or two different DYRK1A shRNAs and stimulated with BAFF. (C) Viable cell quantification by flow cytometry, based on SYTOX blue cell staining, of control BALL1 cells (shControl), DYRK1A-knockdown (using DYRK1A shRNA #1) BALL1 cells (shDYRK1A), or DYRK1A-knockdown BALL1 cells and transduced with the NIK expression vector (shDYRK1A + NIK), cultured in the presence of BAFF or medium control. Data are mean ± SD values combined from the mean values of 3 independent experiments. (D) Flow cytometric analysis of viable and dead cells, based on SYTOX blue cell staining, in BALL1 cells or BALL1 cells transduced with a NIK expression vector (BALL1-NIK). The cells were cultured for 8 days in the presence of BAFF or medium control along with the DYRK1a inhibitor EHT1610 (10 µM) or DMSO. The summary graph is based on 3 replicate samples of the same experiment. (E) Immunoblot analysis of p100 and its processing product p52 in BALL1 or BALL1-NIK cells cultured for 8 days in the presence (+) or absence (–) of BAFF and EHT1610. Summary graph presents p52/p100 ratio based on densitometric quantification of immunoblot protein bands in 3 independent experiments. P values are determined by an unpaired, 2-tailed Student t test (D-E) or 2-way ANOVA with Bonferroni correction (C). *P < .05; **P < .01; ***P < .001.
We next examined whether pharmacological inhibition of DYRK1a interferes with B-ALL cell survival. Interestingly, BAFF-induced BALL1 cell survival was largely impaired in the presence of a selective DYRK1a inhibitor, EHT1610,35 causing a drastic increase in cell death (Figure 6D). Treatment of BALL1 cells with EHT1610 also promoted BALL1 cell death under steady-state conditions, albeit less strikingly than under BAFF-induced conditions (Figure 6D). Importantly, ectopic expression of NIK in BALL1 cells rendered these B-ALL cells largely resistant to EHT1610-mediated cell death (Figure 6D). Consistently, EHT1610 potently inhibited BAFF-induced p100 processing, which was rescued by ectopic NIK expression. These results further suggest that DYRK1a promotes B-ALL cell survival via the ncNF-κB signaling pathway.
DYRK1a-NIK signaling axis regulates B-cell leukemogenesis in mice
To further determine the role of the DYRK1a-NIK signaling axis in B-ALL, we used an animal model36 by transplanting lethally irradiated B6 mice with WT or Dyrk1aBKO bone marrow cells infected with retroviruses expressing the oncogene BCR-ABL and GFP marker gene (supplemental Figure 10A). In this case, we generated the Dyrk1aBKO mice by crossing Dyrk1a-flox mice with Mb1-Cre mice known to strongly express the Cre recombinase in early stages of bone marrow B cells.28 Mice transplanted with BCR-ABL–transduced WT bone marrow cells developed B-ALL, characterized by early lethality and the presence of a high frequency of GFP+ leukemic B cells in the blood, bone marrow, and various other organs, but these B-ALL phenotypes were severely attenuated in the recipient mice of Dyrk1aBKO bone marrow cells (supplemental Figure 10B-D). Like the human B-ALL cells, these mouse leukemic cells displayed pre–B-cell surface markers (CD19+, IgM−, and BP-1+) and expressed high levels of BAFFR (Figure 7A; supplemental Figure 10E). Interestingly, freshly isolated WT B-ALL cells displayed potent ncNF-κB activation, which was largely impaired in the Dyrk1aBKO B-ALL cells (Figure 7B). Consistently, the Dyrk1aBKO B-ALL cells had increased apoptosis and decreased proliferative ability compared with the WT B-ALL cells (Figure 7C-D). These findings suggest that DYRK1a-mediated B-ALL development may involve activation of the ncNF-κB pathway.
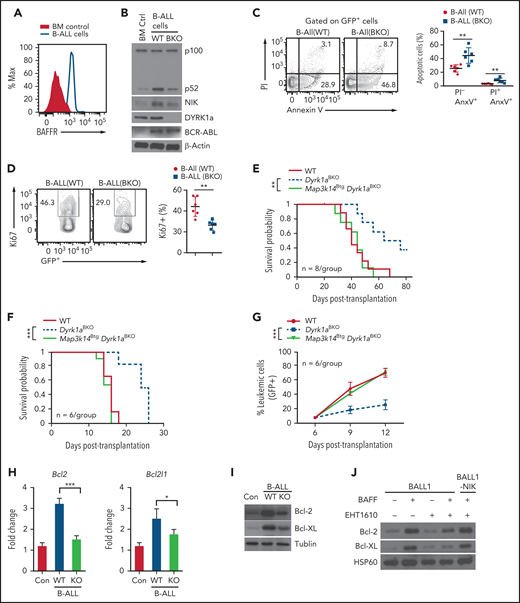
DYRK1a-NIK axis mediates B-ALL induction in a mouse model. (A) Flow cytometric analysis of BAFFR expression using either normal bone marrow control cells or B-ALL cells (GFP+) isolated from B-ALL mice generated by transplanting lethally irradiated C57BL/6 recipients with BCR-ABL-GFP–transduced WT bone marrow cells. (B,I) Immunoblot analysis of the indicated proteins in whole-cell lysates of normal bone marrow control (BM Ctrl) or GFP+ B-ALL cells freshly sorted from B-ALL mice generated using WT or Dyrk1aBKO bone marrow cells (day 40 after bone marrow transplantation). (C) Flow cytometric analysis of apoptotic cells based on annexin V and propidium iodide staining in freshly isolated WT and Dyrk1aBKO B-ALL cells (GFP+). (D) Flow cytometric analysis of proliferating cells (Ki67+) in freshly isolated WT and Dyrk1aBKO B-ALL cells (GFP+). (E) Survival curve of primary B-ALL mice generated with BCR-ABL-GFP–transduced bone marrow cells from wild-type, Dyrk1aBKO, or Map3k14BtgDyrk1aBKO mice. (F,G) Survival curve (I) and frequency of leukemic cells (GFP+) in the peripheral blood lymphocytes (J) of secondary B-ALL mice generated by transplanting sublethally irradiated C57BL/6 recipients with B-ALL cells sorted from the indicated primary B-ALL mice. (H) qRT-PCR analysis of Bcl-2 and Bcl-XL expression using either normal bone marrow control cells or B-ALL cells (GFP+) freshly isolated from B-ALL mice. (J) Immunoblot analysis of Bcl-2 and Bcl-XL in BALL1 or BALL1-NIK cells cultured for 8 days in the presence (+) or absence (–) of BAFF and EHT1610. Data are representative of 2 independent experiments. Summary graphs are mean ± SD values based on multiple mice (C-G) or triplicated samples (H), and P values are determined by an unpaired, 2-tailed Student t test (C-D,H), log-rank Mantel-Cox test (E,F), or two-way ANOVA with Bonferroni correction (G). *P < .05; **P < .01; ***P < .001.
DYRK1a-NIK axis mediates B-ALL induction in a mouse model. (A) Flow cytometric analysis of BAFFR expression using either normal bone marrow control cells or B-ALL cells (GFP+) isolated from B-ALL mice generated by transplanting lethally irradiated C57BL/6 recipients with BCR-ABL-GFP–transduced WT bone marrow cells. (B,I) Immunoblot analysis of the indicated proteins in whole-cell lysates of normal bone marrow control (BM Ctrl) or GFP+ B-ALL cells freshly sorted from B-ALL mice generated using WT or Dyrk1aBKO bone marrow cells (day 40 after bone marrow transplantation). (C) Flow cytometric analysis of apoptotic cells based on annexin V and propidium iodide staining in freshly isolated WT and Dyrk1aBKO B-ALL cells (GFP+). (D) Flow cytometric analysis of proliferating cells (Ki67+) in freshly isolated WT and Dyrk1aBKO B-ALL cells (GFP+). (E) Survival curve of primary B-ALL mice generated with BCR-ABL-GFP–transduced bone marrow cells from wild-type, Dyrk1aBKO, or Map3k14BtgDyrk1aBKO mice. (F,G) Survival curve (I) and frequency of leukemic cells (GFP+) in the peripheral blood lymphocytes (J) of secondary B-ALL mice generated by transplanting sublethally irradiated C57BL/6 recipients with B-ALL cells sorted from the indicated primary B-ALL mice. (H) qRT-PCR analysis of Bcl-2 and Bcl-XL expression using either normal bone marrow control cells or B-ALL cells (GFP+) freshly isolated from B-ALL mice. (J) Immunoblot analysis of Bcl-2 and Bcl-XL in BALL1 or BALL1-NIK cells cultured for 8 days in the presence (+) or absence (–) of BAFF and EHT1610. Data are representative of 2 independent experiments. Summary graphs are mean ± SD values based on multiple mice (C-G) or triplicated samples (H), and P values are determined by an unpaired, 2-tailed Student t test (C-D,H), log-rank Mantel-Cox test (E,F), or two-way ANOVA with Bonferroni correction (G). *P < .05; **P < .01; ***P < .001.
To further determine the role of the ncNF-κB pathway in DYRK1a-mediated B-ALL induction, we tested whether transgenic expression of NIK in the Dyrk1aBKO bone marrow B cells is able to rescue their defect in leukemogenesis. For these studies, we performed B-ALL using BCR-ABL–transduced bone marrow cells derived from the wild-type, Dyrk1aBKO, and Dyrk1aBKOMap3k14Btg mice. While mice transplanted with the Dyrk1aBKO bone marrow cells displayed improved survival, this phenotype was completely reversed in mice transplanted with the Dyrk1aBKOMap3k14Btg bone marrow cells (Figure 7E). To assess the role of the DYRK1a-NIK signaling axis in the survival and expansion of the B-ALL cells, we performed secondary B-ALL by injecting primary B-ALL cells to lethally irradiated B6 mice. As expected,36 induction of secondary B-ALL occurred rapidly, and all mice receiving the WT primary leukemic cells died within 16 days (Figure 7F). Moreover, mice receiving the Dyrk1aBKO primary leukemic cells had profoundly improved survival and reduced leukemic cell frequency (Figure 7F-G). Importantly, ectopic NIK expression in the Dyrk1aBKO primary leukemic cells completely restored their ability to efficiently induce secondary B-ALL (Figure 7F-G). Collectively, these findings suggest a critical role for the DYRK1a-NIK signaling axis in regulating B-ALL development.
To assess the molecular mechanism by which DYRK1a-NIK axis regulates the survival of B cells and B-ALL cells, we analyzed the expression of a panel of anti-apoptotic and pro-apoptotic genes from the Bcl2 family by quantitative reverse transcription polymerase chain reaction (qRT-PCR). We identified two anti-apoptotic genes, Bcl2 and Bcl2l1 (Bcl-XL), which was induced by BAFF in splenic B cells in a Dyrk1a-dependent manner (supplemental Figure 11A). Bcl2 and Bcl2l1 were also highly expressed in B-ALL cells (compared with normal bone marrow cells), and the expression of these survival genes was significantly reduced in the Dyrk1aBKO B-ALL cells (Figure 7H). The protein level expression of Bcl-2 and Bcl-XL was also substantially reduced in the Dyrk1aBKO B-ALL cells (Figure 7I). Moreover, pharmacological inhibition of DYRK1a also attenuated BAFF-stimulated expression of Bcl-2 and Bcl-XL in the human B-ALL cell line BALL1, which could be rescued by ectopic NIK expression (Figure 7J). By analyzing a human pediatric ALL dataset, we found that the expression level of Bcl-XL, as well as of DYRK1A, is inversely correlated with patient survival (supplemental Figure 11B). These results provide mechanistic insight into the function of DYRK1a-NIK signaling axis in B-ALL cell survival.
Discussion
Data presented in this paper establish DYRK1a as a kinase that responds to the ncNF-κB inducers BAFF and CD40L and mediates peripheral B-cell survival and maturation. DYRK1a deficiency causes a profound reduction in the mature and T2 immature B-cell populations, which are known to rely on BAFF for survival. We identified DYRK1a as a TRAF3-binding protein required for BAFF- and CD40L-induced ncNF-κB activation and B-cell survival.
It is generally thought that TRAF3 functions as an adaptor recruiting NIK to the cIAP-TRAF E3 ligase complex for ubiquitination and proteolysis.14,15 However, how the receptor signals might trigger modifications of the cIAP-TRAF components leading to NIK stabilization has remained unclear. Our data suggest that the kinase DYRK1a responds to BAFF and CD40L signals and mediates phosphorylation of TRAF3 at S-29. This residue is conserved across the species and located adjacently to the RING domain of TRAF3 known to be essential for its function in the regulation of ncNF-κB signaling.37 It is thus possible that the DYRK1a-mediated TRAF3 phosphorylation may interfere with its function, thereby facilitating NIK accumulation and ncNF-κB activation.
A previous study suggests that DYRK1a deletion in hematopoietic progenitor cells using the interferon-inducible Mx1-Cre causes a defect in lymphopoiesis, leading to reduced frequency of pre-B cells.29 In support of this previous work, we found that DYRK1a deletion using Mb1-Cre, which efficiently expresses the Cre recombinase in early stages of bone marrow B cells,28 results in reduced frequency of pre-B cells. However, this early-stage B-cell defect was not seen in Dyrk1aBKO mice generated using the Cd19-Cre, likely because of the predominant Cre recombinase expression in peripheral, but not bone marrow, B cells.28 Using the Cd19-Cre Dyrk1aBKO mice, we demonstrated a crucial role of DYRK1a in peripheral B-cell maturation and maintenance. Our data suggest that DYRK1a also plays a role in regulating autoimmune B-cell activation and antibody production in the BM12 model of SLE. The requirement of DYRK1a in T-cell–mediated autoimmune B-cell activation is in line with our finding that DYRK1a also mediates ncNF-κB activation by CD40, a crucial receptor involved in T-cell–dependent B-cell activation.38
B-ALL represents the most common form of childhood cancer.39 Despite the substantial advancement in B-ALL treatment, it fails in up to 20% of patients, emphasizing the importance for characterizing additional drug targets. Human DYRK1a gene locus amplification is associated with Down syndrome and B-ALL, and a role for DYRK1a in regulating B-ALL has also been suggested in a mouse model study.20,40,41 Our analysis of a human pediatric ALL database revealed DYRK1a amplification or mRNA overexpression in 10.83% of total cases. Integration of DYRK1a inhibitor into the treatment of DYRK1ahi ALL population may improve the outcomes. Better understanding of the mechanism of DYRK1a function will create additional therapeutic opportunities. Our present study suggests a crucial role of DYRK1a in mediating the BAFFR survival pathway. Notably, although normal B cells do not express BAFFR in their early stages of development, B-ALL cells are characterized by high levels of BAFFR expression despite pre–B-cell origin.21 We found that DYRK1a mediates BAFF-induced ncNF-κB activation and survival of human B-ALL cells. The survival defect of the DYRK1a-knockdown B-ALL cells could be rescued by ectopic expression of NIK. Furthermore, leukemia cells derived from the mouse B-ALL model also express BAFFR, and the defect of Dyrk1aBKO mice in B-ALL induction could be rescued by transgenic expression of NIK. These findings suggest that mediating ncNF-κB signaling is an important mechanism by which DYRK1a regulates peripheral B-cell survival and B-ALL pathogenesis.
Acknowledgments
The authors thank Kamon Sanada for DYRK1a expression vectors, as well as the personnel from the flow cytometry, advanced microscopy, DNA analysis, and animal facilities at The University of Texas MD Anderson Cancer Center for their technical assistance.
This study was supported by grants from the National Institutes of Health, National Institute of General Medical Sciences (GM84459). The Baylor College of Medicine Mass Spectrometry Proteomics Core is supported by the Dan L. Duncan Comprehensive Cancer Center NIH Award (P30 CA125123) and CPRIT Core Facility Award (RP170005). T.G. was a visiting student supported by a scholarship from the China Scholarship Council with the grant number of 201906380080.
Authorship
Contribution: Y.L. designed and performed the research, prepared the figures, and wrote the manuscript; X.X., Z.J., L.Z., J.-Y.Y., C.-J.K., T.G., and X.C. contributed experiments; A.J. and S.Y.J. performed MS and analyzed the MS data; N.B. and M.Y.K. contributed key reagents; and S.-C.S. supervised the work and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Shao-Cong Sun, The University of Texas MD Anderson Cancer Center, 7455 Fannin St, Unit 902, Houston, TX 77030; e-mail: ssun@mdanderson.org.
For original data, please contact ssun@mdanderson.org.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal