

Key Points
Platelet reactivity and platelet count interact biologically with high VWF plasma levels, resulting in an increased risk for VTE.

Abstract
Plasma von Willebrand factor (VWF) and platelet reactivity are risk factors for venous thromboembolism (VTE), and VWF can promote hemostasis by interaction with platelets. In this study, we explored the combined effects of plasma VWF and platelet measures on the risk of incident VTE. A population-based nested case-control study with 403 cases and 816 controls was derived from the Tromsø Study. VWF, platelet count and mean platelet volume (MPV) were measured in blood samples drawn at baseline. Odds ratios (ORs) with 95% confidence intervals (CIs) for VTE were estimated across VWF tertiles, within predefined MPV (<8.5, 8.5-9.5, and ≥9.5 fL) and platelet count (<230, 230-299, and ≥300 ×109/L) strata. Here, participants with VWF levels in the highest tertile and with MPV ≥9.5 fL had an OR of 1.98 (95% CI, 1.17-3.36) for VTE compared with those in the lowest VWF tertile and with MPV <8.5 fL in the age- and sex-adjusted model. In the joint exposure group, 48% (95% CI, 15-96) of VTEs were attributable to the biological interaction between VWF and MPV. Similarly, individuals with VWF in the highest tertile and platelet count ≥300 × 109/L had an OR of 2.91 (95% CI, 1.49-5.67) compared with those with VWF in the lowest tertile and platelet count <230 × 109/L, and 39% (95% CI, −2 to 97) of VTEs in the joint exposure group were explained by the interaction. Our results suggest that platelet reactivity and platelet count interact biologically with high plasma VWF, resulting in an increased risk for incident VTE.
Introduction
Venous thromboembolism (VTE), encompassing deep vein thrombosis (DVT) and pulmonary embolism (PE), is a common disease associated with severe complications.1,2 The incidence of VTE is increasing, contributing to a substantial disease burden globally.3,4 Although the pathophysiology of VTE has gradually been unraveled over the last decades, up to half of all events arise in the absence of any known attributable factors.5,6 Therefore, it is essential to identify risk markers and expand our insight into the mechanisms involved in the development of disease.
von Willebrand Factor (VWF) is a multimeric glycoprotein that is involved in hemostasis. It is synthesized by endothelial cells and megakaryocytes, and stored and released from Weibel-Palade bodies and platelet α-granules upon stimulation.7-9 VWF has 2 distinct roles in hemostasis: to serve as the carrier and protector of coagulation factor VIII (FVIII) and to promote adhesion and aggregation of platelets through interaction with their glycoproteins.10 We and other investigators have demonstrated that elevated plasma levels of VWF are associated with the risk of VTE.11-13 Moreover, a recent Mendelian randomization analysis indicated a causal role for VWF in the development of VTE, but this causality could not be dissected from FVIII because of the tight correlation.14 It is a common notion that the increased VTE risk by elevated VWF levels is mediated primarily through the parallel increase in FVIII levels.12,14,15 However, studies in mice have suggested that VWF-mediated platelet adhesion has a critical role in VTE formation independent of FVIII.16,17
Platelets constitute a crucial part of hemostasis, and have been connected with thrombogenesis through several mechanisms.18,19 A high platelet count is a marker of VTE risk in cancer patients, but not in cancer-free subjects.20,21 Mean platelet volume (MPV), an indirect marker of platelet reactivity,22,23 is associated with the future risk of VTE and unprovoked events, in particular.24 However, studies using other measures of platelet function have provided conflicting results,25 and the association between platelet function and VTE remains insufficiently understood.
Expanded knowledge on the role of the VWF-platelet interaction in the pathogenesis of VTE may facilitate development of novel therapeutic targets and improve strategies for prevention. To focus on the FVIII-independent role of VWF, our aim was to explore the combined effects of categories of VWF levels and platelet measures on the risk of VTE. To address this question, we carried out a nested case-control study derived from a population-based cohort, with the hypothesis that VWF levels and platelet measures would have more than an additive effect on VTE risk.
Methods
Study design
The Tromsø Study is a single-center, population-based cohort with repeated health surveys of the inhabitants of Tromsø municipality in Norway.26 The fourth survey (Tromsø 4) was conducted in 1994 to 1995. All inhabitants aged ≥25 years were invited, and 27 158 individuals (77% of the eligible) took part in the survey. All participants were followed from the date of inclusion until an incident VTE, death, migration, or the end of follow-up (1 September 2007).
During follow-up, 462 participants developed an incident VTE event. For each case, 2 age- and sex-matched controls, who were alive at the index date of the case, were randomly sampled from the source cohort (Figure 1). The age matching was based on the same year of birth. Because of the insufficient quality of plasma samples, 59 VTE cases and 108 controls were excluded, leaving 403 cases and 816 controls in the final sample. Written informed consent was provided by all participants, and study approval was obtained from the regional committee for medical and health research ethics.
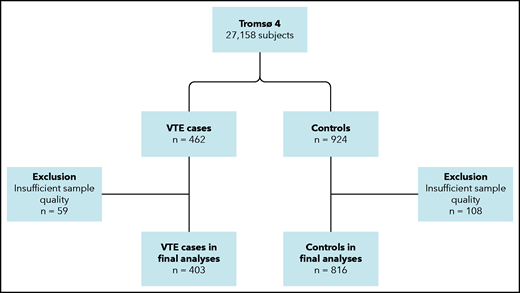
Flowchart of the study population. The chart illustrates the nested case-control design. Subjects (aged ≥25 years) were recruited from the general population. Cases and controls were matched on age and sex.
Flowchart of the study population. The chart illustrates the nested case-control design. Subjects (aged ≥25 years) were recruited from the general population. Cases and controls were matched on age and sex.
Validation of events
To identify all first lifetime VTE events, we searched the hospital discharge diagnosis registry, the radiology procedure registry, and the autopsy registry from the University Hospital of North Norway, which is the only hospital in the study region. The medical record of every patient with a potential VTE was reviewed by trained personnel, and the event was only recorded when clinical signs and symptoms were followed by objective radiological confirmation, resulting in a VTE diagnosis requiring treatment (unless contraindications were specified).
All events were classified as a DVT or PE, with simultaneous evidence of both conditions classified as a PE. Further classification into provoked and unprovoked events was also performed. If the patient had ≥1 provoking factor closely preceding the event, it was categorized as provoked. Provoking factors included trauma or surgery within 8 weeks prior to the event, an acute medical condition (myocardial infarction, ischemic stroke, or infectious disease), active cancer, and immobilization (>3 days’ bed rest, wheelchair confinement, or long-distance travel ≥4 hours within the last 14 days). If the treating physician specified another factor to have provoked the event (eg, venous catheters), this was also recognized.
Baseline measurements and blood sampling
Baseline information was collected by physical examinations, self-administered questionnaires, and blood samples. Height (to the nearest centimeter) and weight (to the nearest 0.5 kg) were measured with participants in light clothing and no shoes; body mass index (BMI) was calculated as weight in kilograms divided by the square of height in meters. Trained personnel performed 3 consecutive blood pressure measurements on all participants, using an automatic device (Dinamap Vital Signs Monitor). Participants rested in a sitting position for 2 minutes before and between the measurements; the last 2 measurements were used to calculate mean systolic and diastolic blood pressure. Self-administered questionnaires were used to obtain information regarding smoking habits and estrogen use, as well as history of cancer and cardiovascular disease (myocardial infarction, stroke, or angina pectoris).
Nonfasting blood samples were collected from an antecubital vein into 5-mL Vacutainers (Becton Dickinson, Le Pont-de-Claix, France) with EDTA (K3-EDTA 40 µL, 0.37 mol/L per tube) as anticoagulant. Platelet-poor plasma was prepared by centrifugation at 3000g at room temperature for 10 minutes. The supernatant was transferred into cryovials (Greiner Labortechnik, Nürtingen, Germany) in 1-mL aliquots and stored at −80°C until further analysis.
Measurement of platelet count, MPV, and VWF
Platelet count and MPV were analyzed within 12 hours of blood sampling, using an automated blood cell counter (Coulter counter; Coulter Electronics, Luton, United Kingdom).
Measurement of VWF was performed at the Research Institute of Internal Medicine, Oslo University Hospital Rikshospitalet. Stored plasma samples were thawed in a water bath at 37°C for 5 minutes and then centrifuged at 13 500g for 2 minutes to obtain platelet-free plasma. Plasma VWF levels were measured by enzyme immunoassays with antibodies (A0082, P02256) obtained from Dako (Glostrup, Denmark), using a polyclonal antibody for coat (A0082) and a horseradish peroxidase–conjugated polyclonal antibody for detection (P02256). Parallel diluted pooled human plasma from 20 healthy individuals was used as standard, and the measurements were expressed as a percentage of the control population mean (100%). The intra- and interassay coefficients of variation were 2.6% and 10.8%, respectively.
Statistical analyses
STATA version 16.0 (Stata Corporation, College Station, TX) and R version 3.6.3 (The R Foundation for Statistical Computing, Vienna, Austria) were used to carry out the statistical analyses.
Categories of VWF (low, medium, high) were defined according to tertiles of plasma VWF levels in the control group. Categories of platelet variables were made with cutoffs at 8.5 and 9.5 fL for MPV and 230 and 300 × 109/L for platelet count, according to our previous study on platelets and VTE risk.24 The 3-level variables of VWF and MPV were combined to yield a 9-level variable; subjects with concomitant low VWF levels and low MPV served as the reference group. A combined category of VWF and platelet count was created using the same approach.
Baseline characteristics across tertiles of VWF levels were expressed as mean (± standard deviation) or median (25th to 75th percentile) for continuous variables and as percentages (quantity) for categorical variables. Unconditional logistic regression was used to estimate odds ratios (ORs) with 95% confidence intervals (CIs) for VTE according to combined categories of VWF and platelet measures. The ORs were adjusted for age and sex in model 1 to take into account the matching factors in the analyses,27 with further adjustment for BMI, C-reactive protein (CRP), smoking, hypertension, estrogen use, self-reported history of cancer at baseline, and platelet count/MPV in model 2.
The relative excess risk due to interaction (RERI)28 was used to evaluate biological interaction: RERI = (ORAB − 1) − (ORA + ORB − 2), where the exposures A and B represent high plasma VWF and high platelet measures, respectively. RERI >0 indicates that the combined effects of the 2 exposures exceed the sum of the individual effects, thereby suggesting biological interaction. The attributable proportion (AP; ie, the proportion of events in the joint exposure group that could be attributed to the biological interaction) was estimated as RERI/ORAB.28 RERI and AP were presented with 95% CIs.28
Results
The baseline characteristics of study participants across tertiles of plasma VWF levels are shown in Table 1. The mean age and proportion with hypertension increased, whereas platelet count decreased slightly, across tertiles of VWF. In the lowest tertile, history of cardiovascular disease was less frequent, whereas smoking was more common. Mean MPV, mean BMI, median CRP, proportion of men/women, estrogen use, and history of cancer did not differ across tertiles of VWF. Median time from baseline to VTE event was 7.5 years. The characteristics of the 403 VTE events are presented in Table 2. The mean age of the patients at the time of their first VTE was 67 years, and 48% were male. DVT (62%) was more common than PE (38%), and 58% of all events were classified as provoked.
Distribution of baseline characteristics according to tertiles of plasma levels of VWF
| Plasma VWF, % | |||
|---|---|---|---|
| Tertile 1 (< 90.4) | Tertile 2 (90.4-100.9) | Tertile 3 (≥100.9) | |
| N | 387 | 410 | 422 |
| Platelet count, mean ± SD, ×109/L | 251 ± 54 | 245 ± 53 | 238 ± 55 |
| MPV, mean ± SD, fL | 8.8 ± 0.9 | 8.8 ± 0.9 | 8.9 ± 1.1 |
| Age, mean ± SD, y | 57.0 ± 14.3 | 60.0 ± 13.7 | 63.4 ± 12.9 |
| Males | 47.8 (185) | 47.3 (194) | 45.7 (193) |
| BMI, mean ± SD, kg/m2 | 25.8 ± 3.9 | 26.6 ± 4.3 | 26.8 ± 4.6 |
| CRP, median (IQR), mg/L | 1.18 (0.65-2.23) | 1.14 (0.63-1.89) | 1.31 (0.65-2.33) |
| Hypertension* | 49.2 (190) | 56.5 (231) | 60.7 (256) |
| Cancer† | 5.0 (16) | 6.6 (21) | 5.4 (17) |
| CVD† | 12.9 (50) | 17.3 (71) | 18.0 (76) |
| Estrogen use‡ | 4.7 (18) | 5.1 (21) | 4.3 (18) |
| Smoking‡ | 41.1 (159) | 25.1 (103) | 26.1 (110) |
| Plasma VWF, % | |||
|---|---|---|---|
| Tertile 1 (< 90.4) | Tertile 2 (90.4-100.9) | Tertile 3 (≥100.9) | |
| N | 387 | 410 | 422 |
| Platelet count, mean ± SD, ×109/L | 251 ± 54 | 245 ± 53 | 238 ± 55 |
| MPV, mean ± SD, fL | 8.8 ± 0.9 | 8.8 ± 0.9 | 8.9 ± 1.1 |
| Age, mean ± SD, y | 57.0 ± 14.3 | 60.0 ± 13.7 | 63.4 ± 12.9 |
| Males | 47.8 (185) | 47.3 (194) | 45.7 (193) |
| BMI, mean ± SD, kg/m2 | 25.8 ± 3.9 | 26.6 ± 4.3 | 26.8 ± 4.6 |
| CRP, median (IQR), mg/L | 1.18 (0.65-2.23) | 1.14 (0.63-1.89) | 1.31 (0.65-2.33) |
| Hypertension* | 49.2 (190) | 56.5 (231) | 60.7 (256) |
| Cancer† | 5.0 (16) | 6.6 (21) | 5.4 (17) |
| CVD† | 12.9 (50) | 17.3 (71) | 18.0 (76) |
| Estrogen use‡ | 4.7 (18) | 5.1 (21) | 4.3 (18) |
| Smoking‡ | 41.1 (159) | 25.1 (103) | 26.1 (110) |
Unless otherwise noted, data are % (n).
CVD, cardiovascular disease; IQR, interquartile range; SD, standard deviation.
Defined as systolic blood pressure ≥140 mm Hg or diastolic blood pressure ≥90 mm Hg.
Self-reported history of cancer or myocardial infarction, angina, or stroke at baseline.
Self-reported daily use of oral contraceptives or hormonal replacement therapy (estrogen use) or smoking of cigarettes or cigars (smoking).
Characteristics of VTE events (n = 403)
| Characteristics | Data |
|---|---|
| Age at VTE, mean ± SD, y | 67.4 ± 13.7 |
| Sex, male | 48.1 (194) |
| Deep vein thrombosis | 62.3 (251) |
| Pulmonary embolism | 37.7 (152) |
| Unprovoked VTE | 41.9 (169) |
| Provoked VTE | 58.1 (234) |
| Surgery/trauma | 22.3 (90) |
| Acute medical condition | 15.6 (63) |
| Active cancer | 21.8 (88) |
| Immobilization | 18.1 (73) |
| Other* | 4.0 (16) |
| Characteristics | Data |
|---|---|
| Age at VTE, mean ± SD, y | 67.4 ± 13.7 |
| Sex, male | 48.1 (194) |
| Deep vein thrombosis | 62.3 (251) |
| Pulmonary embolism | 37.7 (152) |
| Unprovoked VTE | 41.9 (169) |
| Provoked VTE | 58.1 (234) |
| Surgery/trauma | 22.3 (90) |
| Acute medical condition | 15.6 (63) |
| Active cancer | 21.8 (88) |
| Immobilization | 18.1 (73) |
| Other* | 4.0 (16) |
Unless otherwise noted, data are % (n).
SD, standard deviation
Other factor specified by treating physician to have provoked the event (eg, intravascular catheter).
The ORs for VTE by VWF levels within each MPV stratum and by the combined categories of VWF and MPV are displayed in Figure 2A and Table 3. In participants with low MPV (<8.5 fL), those with high VWF levels had an age- and sex-adjusted OR of 1.31 (95% CI, 0.80-2.17) for VTE compared with those with a low VWF level. In participants with high MPV (≥9.5 fL), high VWF levels yielded an OR of 2.18 (95% CI, 1.15-4.16) for VTE compared with those with low VWF. Compared with those with low MPV and low VWF levels, participants with high MPV and high VWF levels had an OR of 1.98 (95% CI, 1.17-3.36) in analyses adjusted for age and sex. The risk estimates were only modestly affected by multivariable adjustment (Figure 2A; Table 3). Interaction analyses yielded a RERI of 1.00 (95% CI, −0.04 to 2.03); by calculating the AP, we found that 48% (95% CI, 15-96) of the cases in the joint exposure group could be attributed to the biological interaction between VWF and MPV.
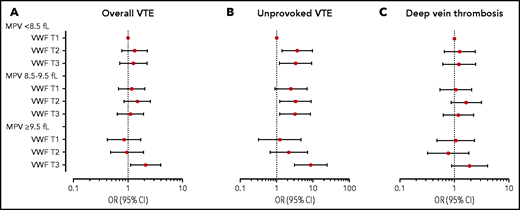
Forest plot of VTE risk by MPV and VWF levels. ORs and 95% CIs in multivariable analyses for overall VTE (A), unprovoked VTE (B), and DVT (C) by categories of VWF and MPV. The group with VWF level in the lowest tertile (T) and low MPV (<8.5 fL) is set as reference. ORs were adjusted for age, sex, BMI, CRP, smoking, hypertension, estrogen use, self-reported history of cancer at baseline, and platelet count.
Forest plot of VTE risk by MPV and VWF levels. ORs and 95% CIs in multivariable analyses for overall VTE (A), unprovoked VTE (B), and DVT (C) by categories of VWF and MPV. The group with VWF level in the lowest tertile (T) and low MPV (<8.5 fL) is set as reference. ORs were adjusted for age, sex, BMI, CRP, smoking, hypertension, estrogen use, self-reported history of cancer at baseline, and platelet count.
ORs with 95% CIs for VTE across tertiles of plasma VWF and categories of MPV
| MPV, fL | VWF, % | Controls (n = 816) | Cases (n = 403) | Within stratum, OR (95% CI) | Combined effects, OR (95% CI) | ||
|---|---|---|---|---|---|---|---|
| Model 1* | Model 2† | Model 1* | Model 2† | ||||
| <8.5 | 314 | 148 | |||||
| T1 | 112 | 46 | Ref | Ref | Ref | Ref | |
| T2 | 106 | 54 | 1.27 (0.79-2.05) | 1.42 (0.81-2.48) | 1.25 (0.78-2.01) | 1.32 (0.77-2.27) | |
| T3 | 96 | 48 | 1.31 (0.80-2.17) | 1.40 (0.77-2.54) | 1.24 (0.76-2.03) | 1.25 (0.70-2.24) | |
| P for trend | .3 | .3 | |||||
| 8.5-9.5 | 324 | 163 | |||||
| T1 | 99 | 47 | Ref | Ref | 1.16 (0.71-1.90) | 1.16 (0.67-2.02) | |
| T2 | 107 | 57 | 1.12 (0.70-1.81) | 1.33 (0.77-2.27) | 1.31 (0.82-2.11) | 1.48 (0.85-2.58) | |
| T3 | 118 | 59 | 1.05 (0.65-1.69) | 0.97 (0.57-1.65) | 1.24 (0.77-1.98) | 1.11 (0.64-1.91) | |
| P for trend | .9 | .9 | |||||
| ≥9.5 | 178 | 92 | |||||
| T1 | 62 | 21 | Ref | Ref | 0.84 (0.46-1.53) | 0.85 (0.42-1.72) | |
| T2 | 60 | 26 | 1.24 (0.63-2.45) | 1.08 (0.49-2.41) | 1.07 (0.60-1.90) | 0.95 (0.48-1.90) | |
| T3 | 56 | 45 | 2.18 (1.15-4.16) | 2.16 (1.01-4.60) | 1.98 (1.17-3.36) | 2.10 (1.11-3.98) | |
| P for trend | .014 | .041 | |||||
| MPV, fL | VWF, % | Controls (n = 816) | Cases (n = 403) | Within stratum, OR (95% CI) | Combined effects, OR (95% CI) | ||
|---|---|---|---|---|---|---|---|
| Model 1* | Model 2† | Model 1* | Model 2† | ||||
| <8.5 | 314 | 148 | |||||
| T1 | 112 | 46 | Ref | Ref | Ref | Ref | |
| T2 | 106 | 54 | 1.27 (0.79-2.05) | 1.42 (0.81-2.48) | 1.25 (0.78-2.01) | 1.32 (0.77-2.27) | |
| T3 | 96 | 48 | 1.31 (0.80-2.17) | 1.40 (0.77-2.54) | 1.24 (0.76-2.03) | 1.25 (0.70-2.24) | |
| P for trend | .3 | .3 | |||||
| 8.5-9.5 | 324 | 163 | |||||
| T1 | 99 | 47 | Ref | Ref | 1.16 (0.71-1.90) | 1.16 (0.67-2.02) | |
| T2 | 107 | 57 | 1.12 (0.70-1.81) | 1.33 (0.77-2.27) | 1.31 (0.82-2.11) | 1.48 (0.85-2.58) | |
| T3 | 118 | 59 | 1.05 (0.65-1.69) | 0.97 (0.57-1.65) | 1.24 (0.77-1.98) | 1.11 (0.64-1.91) | |
| P for trend | .9 | .9 | |||||
| ≥9.5 | 178 | 92 | |||||
| T1 | 62 | 21 | Ref | Ref | 0.84 (0.46-1.53) | 0.85 (0.42-1.72) | |
| T2 | 60 | 26 | 1.24 (0.63-2.45) | 1.08 (0.49-2.41) | 1.07 (0.60-1.90) | 0.95 (0.48-1.90) | |
| T3 | 56 | 45 | 2.18 (1.15-4.16) | 2.16 (1.01-4.60) | 1.98 (1.17-3.36) | 2.10 (1.11-3.98) | |
| P for trend | .014 | .041 | |||||
Ref, reference; T, tertile.
Adjusted for age and sex.
Adjusted for age, sex, BMI, CRP, smoking, hypertension, estrogen use, self-reported history of cancer at baseline, and platelet count.
Subgroup analyses showed that the combined effect of VWF and MPV was particularly strong for unprovoked VTE events; participants with high VWF levels and MPV had an age- and sex-adjusted OR of 6.63 (95% CI, 2.89-15.17) compared with those with low MPV and low VWF levels. Multivariable adjusted analyses yielded similar risk estimates (Figure 2B). The RERI was 5.18 (95% CI, −2.29 to 9.93), and the AP implied that 59% (95% CI, 6-96) of the unprovoked VTEs in the joint exposure group were attributable to this interaction. Stratified analyses further pointed toward a stronger association for DVT (Figure 2C) than for PE; joint exposure of high VWF levels and high MPV yielded age- and sex-adjusted ORs of 2.23 (95% CI, 1.21-4.11) for DVT and 1.61 (95% CI, 0.75-3.46) for PE. RERI was 0.62 (95% CI, −0.50 to 1.83), and the AP suggested that 33% (95% CI, −16 to 99) of the DVT cases in the joint exposure group could be attributed to the interaction between VWF and MPV.
The ORs for VTE across combined categories of plasma VWF level and platelet count are shown in Figure 3A and Table 4. Table 4 also depicts the ORs for VTE according to VWF levels within each stratum of platelet count. In the low platelet count stratum, a high VWF level yielded an age- and sex-adjusted OR of 1.52 (95% CI, 0.95-2.44) compared with those with a low VWF. The corresponding OR in the high platelet count stratum was 2.77 (95% CI, 1.25-6.14). The combination of high platelet count and plasma VWF level in the highest tertile yielded an OR of 2.91 (95% CI, 1.49-5.67) for VTE compared with those with low platelet count and VWF level in the age- and sex-adjusted model. The ORs were minimally affected by further adjustment for BMI, CRP, smoking, hypertension, estrogen use, cancer at baseline, and MPV (Figure 3A; Table 4). Interaction analyses provided a RERI of 0.94 (95% CI, −0.59 to 2.52), indicating that the combined effects of high VWF and high platelet count on VTE risk were more than additive; the AP implied that 39% (95% CI, −2 to 97) of the events in the joint exposure group could be attributed to the interaction.
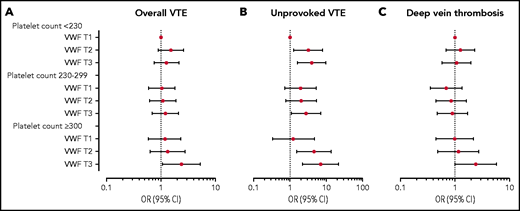
Forest plot of VTE risk by platelet count and VWF levels. ORs and 95% CIs in multivariable analyses for overall VTE (A), unprovoked VTE (B), and DVT (C) by categories of VWF and platelet count. The group with VWF level in the lowest tertile and low platelet count (<230 × 109/L) is set as reference. ORs were adjusted for age, sex, BMI, CRP, smoking, hypertension, estrogen use, self-reported history of cancer at baseline, and MPV. T, tertile.
Forest plot of VTE risk by platelet count and VWF levels. ORs and 95% CIs in multivariable analyses for overall VTE (A), unprovoked VTE (B), and DVT (C) by categories of VWF and platelet count. The group with VWF level in the lowest tertile and low platelet count (<230 × 109/L) is set as reference. ORs were adjusted for age, sex, BMI, CRP, smoking, hypertension, estrogen use, self-reported history of cancer at baseline, and MPV. T, tertile.
ORs with 95% CIs for VTE across tertiles of plasma VWF and categories of platelet count
| Platelet count, ×109/L | VWF, % | Controls (n = 816) | Cases (n = 403) | Within stratum, OR (95% CI) | Combined effects, OR (95% CI) | ||
|---|---|---|---|---|---|---|---|
| Model 1* | Model 2† | Model 1* | Model 2† | ||||
| <230 | 349 | 176 | |||||
| T1 | 112 | 41 | Ref | Ref | Ref | Ref | |
| T2 | 112 | 64 | 1.56 (0.97-2.51) | 1.44 (0.84-2.45) | 1.57 (0.98-2.52) | 1.52 (0.90-2.59) | |
| T3 | 125 | 71 | 1.52 (0.95-2.44) | 1.13 (0.66-1.95) | 1.56 (0.98-2.49) | 1.28 (0.76-2.17) | |
| P for trend | .1 | .7 | |||||
| 230-299 | 349 | 157 | |||||
| T1 | 111 | 49 | Ref | Ref | 1.22 (0.74-1.99) | 1.04 (0.60-1.82) | |
| T2 | 117 | 52 | 1.01 (0.63-1.61) | 1.06 (0.62-1.83) | 1.22 (0.75-1.99) | 1.09 (0.63-1.90) | |
| T3 | 121 | 56 | 1.03 (0.64-1.64) | 1.17 (0.69-2.00) | 1.29 (0.80-2.08) | 1.23 (0.71-2.10) | |
| P for trend | .9 | .6 | |||||
| ≥300 | 118 | 70 | |||||
| T1 | 50 | 24 | Ref | Ref | 1.30 (0.71-2.39) | 1.18 (0.59-2.33) | |
| T2 | 44 | 21 | 1.09 (0.53-2.27) | 1.30 (0.54-3.13) | 1.32 (0.70-2.48) | 1.34 (0.64-2.83) | |
| T3 | 24 | 25 | 2.77 (1.25-6.14) | 2.52 (0.95-6.69) | 2.91 (1.49-5.67) | 2.40 (1.08-5.34) | |
| P for trend | .017 | .073 | |||||
| Platelet count, ×109/L | VWF, % | Controls (n = 816) | Cases (n = 403) | Within stratum, OR (95% CI) | Combined effects, OR (95% CI) | ||
|---|---|---|---|---|---|---|---|
| Model 1* | Model 2† | Model 1* | Model 2† | ||||
| <230 | 349 | 176 | |||||
| T1 | 112 | 41 | Ref | Ref | Ref | Ref | |
| T2 | 112 | 64 | 1.56 (0.97-2.51) | 1.44 (0.84-2.45) | 1.57 (0.98-2.52) | 1.52 (0.90-2.59) | |
| T3 | 125 | 71 | 1.52 (0.95-2.44) | 1.13 (0.66-1.95) | 1.56 (0.98-2.49) | 1.28 (0.76-2.17) | |
| P for trend | .1 | .7 | |||||
| 230-299 | 349 | 157 | |||||
| T1 | 111 | 49 | Ref | Ref | 1.22 (0.74-1.99) | 1.04 (0.60-1.82) | |
| T2 | 117 | 52 | 1.01 (0.63-1.61) | 1.06 (0.62-1.83) | 1.22 (0.75-1.99) | 1.09 (0.63-1.90) | |
| T3 | 121 | 56 | 1.03 (0.64-1.64) | 1.17 (0.69-2.00) | 1.29 (0.80-2.08) | 1.23 (0.71-2.10) | |
| P for trend | .9 | .6 | |||||
| ≥300 | 118 | 70 | |||||
| T1 | 50 | 24 | Ref | Ref | 1.30 (0.71-2.39) | 1.18 (0.59-2.33) | |
| T2 | 44 | 21 | 1.09 (0.53-2.27) | 1.30 (0.54-3.13) | 1.32 (0.70-2.48) | 1.34 (0.64-2.83) | |
| T3 | 24 | 25 | 2.77 (1.25-6.14) | 2.52 (0.95-6.69) | 2.91 (1.49-5.67) | 2.40 (1.08-5.34) | |
| P for trend | .017 | .073 | |||||
T, tertile.
Adjusted for age and sex.
Adjusted for age, sex, BMI, CRP, smoking, hypertension, estrogen use, self-reported history of cancer at baseline, and MPV.
Subgroup analyses of the interaction between VWF and platelet count provided similar results as for MPV, with the strongest associations for unprovoked events and DVT. The combination of a high platelet count and high VWF yielded age-and sex-adjusted ORs of 6.54 (95% CI, 2.47-17.32) for unprovoked VTE and 3.01 (95% CI, 1.45-6.23) for DVT compared with those with low levels in both exposures. Multivariable adjustment did not affect the estimates substantially (Figure 3B-C). RERI for unprovoked VTE was 2.73 (95% CI, −0.93 to 4.36), and the AP indicated that 40% (95% CI, 10-100) of unprovoked VTEs in those with combined high platelet count and high VWF could be attributed to the interaction. For DVT, the RERI was 1.36 (95% CI, −0.68 to 3.54), and the AP implied that 56% (95% CI, 8-100) of all DVTs in the joint exposure group were attributable to the interaction.
Discussion
In the present population-based nested case-control study, we found that the combination of elevated plasma VWF levels and high MPV or a high platelet count had a more than additive effect on the VTE risk. In those with high MPV and high VWF, 48% of the VTE events could be attributed to the biological interaction. Similarly, 39% of the VTE events in those with high platelet count and high VWF could be attributed to the interaction. The combinations of elevated VWF levels and high platelet measures were most strongly associated with unprovoked VTE events. Our findings suggest that the presence of high VWF levels and a high platelet count or reactivity is required to yield an increased risk for VTE, implying a codependency between VWF and platelet measures in the biology of the VTE risk.
MPV is a marker of platelet reactivity and functional capacity,29 because large platelets are observed to have a higher turnover of thromboxane A2 and release larger amounts of signaling substances upon activation.22,30 Platelet size is primarily determined by the state of the parent megakaryocyte at the time of discharge.31,32 In situations with enhanced platelet consumption, such as hypoxia, smoking, high BMI, and immune thrombocytopenic purpura, the megakaryocytes have higher cytoplasmic volumes and release larger platelets.22,33,34 Large and more reactive (and hemostatically active) platelets are a risk factor for arterial35,36 and venous24 thrombosis.
Plasma VWF levels are shown to be associated with future risk of VTE.11,13 VWF is acknowledged as an important protagonist of arterial thrombosis under high-flow conditions,37 whereas the role of VWF in the causal pathway of VTE is still not fully understood. Since the discovery of the association between VWF and DVT in 1995, it has been perceived that it is primarily explained by concurrent high levels of FVIII.12 However, in the Longitudinal Investigation of Thromboembolism Etiology study, VWF and FVIII were reported to be independently associated with the risk of VTE.11 A recent Mendelian randomization analysis was not able to address the independent role of VWF, because no locus was found to regulate VWF independently of FVIII.14 In addition to serving as the carrier of FVIII, VWF promotes hemostasis by interacting with platelet glycoprotein Ib-IX-V and αIIbβ3 integrin, resulting in adhesion of platelets to the endothelium and to other platelets, respectively.38 Therefore, it is reasonable to assume that VWF is also involved in FVIII-independent mechanisms in the pathogenesis of VTE.
To our knowledge, this is the first study to explore the combined effects of VWF with platelet count and platelet reactivity on VTE risk. The observation that VWF and platelet measures displayed a synergistic effect on the risk of VTE suggests that a biological interaction may contribute to the pathophysiology of VTE. Our results add knowledge about the presence and impact of this interaction in the general population. Notably, for overall VTE, VWF and platelet measures only yielded increased VTE risk when both components were elevated. Specifically, the 2 variables did not merely interact to add excess risk39 but appeared to be dependent on each other to mediate increased risk. Thus, codependency, rather than synergism, may be a more fitting term for the interaction.
Similar to previous studies investigating the separate association of VWF and MPV in relation to VTE risk,24 we found that the combined effect was strongest for unprovoked events. This implies a contribution by the VWF-platelet interaction on unprovoked events, in particular.40,41 Furthermore, the combined exposure of high VWF levels and high MPV or high platelet count resulted in a particularly strong increase in the risk for DVT compared with PE. This finding is analogous with our previous study on VWF and the risk for VTE13 and suggests that thrombi formed in the presence of high plasma levels of VWF and either many or highly reactive platelets may be more tightly anchored to the endothelium and, thus, be less likely to embolize.
Evidence from mouse studies suggests an essential role for VWF-platelet interaction in the pathogenesis of VTE. First, Chauhan and colleagues found that VWF-deficient mice had impaired thrombus growth in ferric chloride–injured veins.16 Second, Brill and colleagues reported that the VWF-platelet interaction was necessary for the development of venous thrombosis in a flow-restricted model.17 Traditionally, an increased thrombin potential because of concurrent elevated FVIII levels has been thought to explain the observed VWF-associated VTE risk.12,14 However, infusion of recombinant FVIII in VWF-deficient mice only resulted in increased thrombus stability and did not increase thrombus formation.16,17 Recently, Michels and colleagues found that VWF is critical in obesity-associated DVT in mice and demonstrated that targeting the VWF-platelet interaction with antibodies or nanobodies protected against thrombogenicity.42 Furthermore, observational studies have shown that VWF and FVIII are independent risk factors for VTE.11,43 Taken together, growing evidence suggests that enhanced platelet reactivity contributes to the increased VTE risk in those with elevated plasma VWF levels. The VWF-platelet interaction may therefore represent a promising target for VTE prevention and treatment, as for arterial thrombosis.42,44 Several studies have been carried out on possible therapeutic approaches for VWF inhibition, but the focus has been on arterial thrombosis.45
Platelet count is inversely correlated with MPV and bleeding time22 but is not regarded as a useful marker of a prothrombotic state because no association is established with arterial or venous thrombotic events,24,46,47 and both low and high platelet count are associated with increased mortality and the risk of bleeding events in the general population.48-52 However, the risk of VTE was substantially increased in subjects with high platelet count and concomitant high VWF, suggesting that the VWF-platelet interaction is also important for thrombogenesis in the presence of a high platelet count.
The strengths of this study include the population-based nested case-control design with a large population of cases and controls sampled from the same parent cohort with close follow-up. Because of the prospective design, we were able to gain insight into the temporal sequences of the associations. The study also has some limitations that require attention. Guidelines recommend collecting blood into tubes containing citrate as anticoagulant to measure VWF levels,53 but in the present study, only plasma obtained from samples collected into EDTA was available. Of note, previous studies of healthy volunteers suggested a strong positive correlation between VWF antigen levels measured in EDTA and citrate plasma.54,55 Although participants from this population-based nested case-control study were not necessarily healthy at baseline, it is unlikely that they would have clinically relevant coagulation disorders. Still, because blood samples were collected into EDTA in cases and controls, any discrepancy between true and measured levels of VWF would be nondifferential in relation to VTE status. In addition, blood samples were stored for >20 years between baseline sampling and measurement of VWF and were subjected to 1 additional freeze-thaw cycle before assessment of VWF, which could have introduced a discrepancy between true and measured levels. However, the samples were stored in the same way, for the same duration, and were subjected to the same number of freeze-thaw cycles in cases and controls; potential alterations would be nondifferential with regard to VTE status, thereby introducing a possibility for regression dilution bias and a weakening of the results compared with the true associations. Interaction analyses have inherent statistical limitations because they require dividing the study population into smaller groups.56 Thus, our results on measures of biological interaction and their 95% CIs should be interpreted with caution, especially for the VTE subgroups. The majority of participants in this study were white; therefore, we encourage caution when extrapolating these findings to individuals of other ethnicities.
In summary, we found a synergistic effect of elevated plasma VWF levels and high MPV on the risk for VTE, which was most prominent for unprovoked events. High platelet count also yielded an increased risk for VTE when VWF levels were high. Our findings suggest that the presence of high VWF levels and a high platelet count or reactivity are required to yield an increased risk for VTE. Further research is necessary to disentangle the mechanisms of interaction between VWF and platelet number and reactivity and explore potential targets for VTE prevention and treatment.
Authorship
Contribution: M.S.E. performed statistical analyses, interpreted data, and drafted the manuscript; K.H., S.K.B., and L.H.E. performed statistical analyses, interpreted data, and revised the manuscript; E.-S.H. and V.M.M. interpreted data and revised the manuscript; T.U. and P.A. performed laboratory analyses, interpreted data, and revised the manuscript; and J.-B.H. conceived and designed the study, interpreted data and revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Magnus S. Edvardsen, Department of Clinical Medicine, Thrombosis Research Center, UiT – The Arctic University of Norway, N-9037 Tromsø, Norway; e-mail: magnus.s.edvardsen@uit.no.
Data sharing requests should be sent to Magnus S. Edvardsen (magnus.s.edvardsen@uit.no).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal