

TO THE EDITOR:
The treatment of patients with acute promyelocytic leukemia (APL) can serve as a paradigm for cancer therapy.1 The outcome of this disease, in adults and in children, has significantly improved with the introduction of target-specific agents, such as all-trans retinoic acid (ATRA) and arsenic trioxide (ATO), providing long-term survival for most patients.2,3 Although morphologic and clinical suspicion is sufficient to immediately initiate ATRA, definitive diagnosis relies on the demonstration of PML/RARA translocation, on alternative RARA rearrangements represented by RARA gene fusion to other partners, cryptic insertion into the PML gene, or vice versa that altogether occur in more than 98% of APL cases.4-6 Here we describe an even more challenging situation where all conventional diagnostic approaches failed to detect an oncogenic event associated with the diagnosis of APL.
A 6-year-old girl was admitted to the emergency department with a 3-day history of shoulder pain and fever. On physical examination, she presented with pallor and ecchymosis of the lower limbs. A full blood count showed a hemoglobin concentration of 9.7 g/dL, and leukocyte and platelet counts of 2.8 × 103/μL and 101 × 103/μL, respectively. Coagulation tests showed consumptive coagulopathy with slightly prolonged prothrombin and activated partial thromboplastin times, an increased D-dimer concentration, and hypofibrinogenemia. A peripheral blood smear showed atypical promyelocytes packed with numerous azurophilic granules (Figure 1A), whereas analysis of the bone marrow aspirate demonstrated markedly hypercellular marrow containing 85% abnormal promyelocytes with Auer rods (CD33+, CD13+, CD38+, CD99+, HLA-DRlow), with strong and diffuse reactivity to myeloperoxidase staining. Molecular analysis was negative for PML/RARA fusions, and karyotype analysis did not show t(15;17)(q24;q21). Fluorescence in situ hybridization with RARA break-apart probe confirmed the negativity of all RARA rearrangements and of the cryptic RARA gene insertion. Based on the characteristic morphologic features, the review of the peripheral blood and bone marrow was consistent with a diagnosis of hypergranular (or typical) APL, and therapy with ATRA was immediately started. The patient received treatment combining standard induction acute myeloid leukemia therapy and ATRA, followed by 3 high-dose cytarabine-based courses of consolidation therapy. Complete remission was achieved after the first induction cycle and persisted until the end of treatment.

Morphologic features of the APL case and characterization of the TTMV/RARA chimeric fusion transcript. (A) Morphology of the case of APL, negative for RARA rearrangement. Nuclear size and shape are irregular, whereas the cytoplasm is for the most part occupied by densely packed granules. (B) Average relative expression level from WTS data of RARA exons 2 and 3, and the expressed intronic region is shown for the index case (blue squares) and for another 12 AMLs with known chromosomal aberrations. (C) Schematic representation of the fusion between the TTMV sequence and RARA exon 3. The chimeric transcript shows the conserved TTMV UTR sequence followed by the ORF2, immediately upstream of the short sequence of retained RARA intron 2, in frame with the full RARA exon 3. A new splicing event that joins this part of the retained intron 2 to exon 3 and removes the intervening intronic sequence occurs during splicing. Some representative sequencing reads that support the presence of the fusion transcript are shown. The exon–intron gene structure and the location of the integrated viral sequence is indicated at the bottom. (D-E) Sanger sequencing electropherograms of the downstream (D) and upstream (E) breakpoints of TTMV integration on the leukemia relapse sample genomic DNA. (F) Detection of viral integration with primers common to the 3 Anelloviruses TTV, TTMDV, and TTMV (NG779/NG782), and those specific only for TTMV (NG792/NG791), in genomic DNA of the relapsed APL case and in another 6 AML samples. (G-H) Detection of the TTMV/RARA chimeric fusion in genomic DNA (G) and cDNA (H) in sequential samples of the APL case from diagnosis to relapse, during remission–induction therapy, and after HSCT. (I) Quantitative real-time PCR detection of the TTMV/RARA fusion transcript decrease during induction therapy and after HSCT.
Morphologic features of the APL case and characterization of the TTMV/RARA chimeric fusion transcript. (A) Morphology of the case of APL, negative for RARA rearrangement. Nuclear size and shape are irregular, whereas the cytoplasm is for the most part occupied by densely packed granules. (B) Average relative expression level from WTS data of RARA exons 2 and 3, and the expressed intronic region is shown for the index case (blue squares) and for another 12 AMLs with known chromosomal aberrations. (C) Schematic representation of the fusion between the TTMV sequence and RARA exon 3. The chimeric transcript shows the conserved TTMV UTR sequence followed by the ORF2, immediately upstream of the short sequence of retained RARA intron 2, in frame with the full RARA exon 3. A new splicing event that joins this part of the retained intron 2 to exon 3 and removes the intervening intronic sequence occurs during splicing. Some representative sequencing reads that support the presence of the fusion transcript are shown. The exon–intron gene structure and the location of the integrated viral sequence is indicated at the bottom. (D-E) Sanger sequencing electropherograms of the downstream (D) and upstream (E) breakpoints of TTMV integration on the leukemia relapse sample genomic DNA. (F) Detection of viral integration with primers common to the 3 Anelloviruses TTV, TTMDV, and TTMV (NG779/NG782), and those specific only for TTMV (NG792/NG791), in genomic DNA of the relapsed APL case and in another 6 AML samples. (G-H) Detection of the TTMV/RARA chimeric fusion in genomic DNA (G) and cDNA (H) in sequential samples of the APL case from diagnosis to relapse, during remission–induction therapy, and after HSCT. (I) Quantitative real-time PCR detection of the TTMV/RARA fusion transcript decrease during induction therapy and after HSCT.
Eight months later, the child presented with fever, hemorrhagic findings, and abdominal and muscular pain. Bone marrow aspiration revealed an APL relapse with the same morphologic and immunophenotypic characteristics of the diagnosis, as well as a normal karyotype and the absence of the PML/RARA rearrangement and its isoforms.
Whole-transcriptome sequencing (WTS) of the relapse sample was then carried out, showing the aberrant expression of a small portion of RARA intron 2 (Figure 1B), not belonging to any known isoform and not expressed in a series of other pediatric acute myeloid leukemias (AMLs). The 5' sequence extension from RARA exon 3 revealed the abundant presence of a fusion transcript involving the integration of 209 nucleotides upstream of exon 3, linked by the previously identified short sequence of retained intron 2. The insertion was located at chr17:40334196 and estimated to be 1045 bp in size, of which only the last 209 nucleotides were expressed in the fusion transcript, in-frame with RARA exon 3, and linked by 38 nucleotides of retained intron.
The integrated sequence was not homologous to any human sequence but revealed a significant alignment to different Anelloviridae isolates from torque teno Mini virus (TTMV; 67% to 69% coverage; 74% to 85% identity; Figure 1C-E). It also showed the putative conserved domain of the Torque Teno Open Reading Frame 2 (ORF2) superfamily in the coding region (E value = 3.44e−09), with a protein identity of 48% and a coverage of 72% with respect to TTMV-ORF2. This ORF displays the conserved amino acid motif WX7HX3CXCX5H located at the N terminus, shared among all anelloviruses. The presence of the sequence mapping to the highly conserved Anellovirus untranslated region (UTR) was identified,7,8 specifically the 1 characterizing TTMV isolates,7 only in the index case and not in other 6 AML DNAs (Figure 1F). The full-length TTMV/RARA fusion transcript was predicted to encode a 485-amino-acid protein containing the DNA-binding and ligand-binding domain of RARA. No evidence of the TTMV/RARA fusion sequence was found in AML samples from Leucegene and Therapeutically Applicable Research to Generate Effective Treatments (TARGET) cohorts and in a publicly available dataset of APL, that anyway did not include any case negative for known RARA rearrangement.
The patient received a combination of ATRA and ATO according to emerging evidence9-14 and to the discovery of the novel RARA fusion transcript. Therapy consisting of 2 courses of ATO and ATRA was administered. Complete morphologic remission was achieved on day +21 of the first cycle. After 2 consolidation courses of ATO, given the availability of an HLA-matched sibling donor, she underwent hematopoietic stem cell transplantation (HSCT), resulting in a second complete remission. Bone marrow evaluations performed after HSCT confirmed complete morphologic remission, and analysis of the peripheral blood cells showed stable full donor chimerism. The patient is presently alive and well at 4 years after HSCT. Molecular analysis of leukemic samples before HSCT confirmed the presence of TTMV DNA integration in genomic DNA upstream of RARA exon 3 by polymerase chain reaction (PCR), whereas reverse transcription (RT)-PCR showed the expression of the chimeric fusion in both the diagnosis and the relapse samples (Figure 1G-H). Quantitative RT-PCR revealed the progressive decrease of RARA fusion transcript expression along with remission-induction therapy, with complete clearance of the pathogenic allele after HSCT (Figure 1I).
A retrospective in silico analysis in our WTS database of 22 pediatric cytogenetically normal AML cases identified a second case of TTMV integration upstream of RARA exon 3 in a 3-year-old child affected by an AML lacking any cytogenetic abnormality or pathogenic somatic mutation. The integrated TTMV sequence was highly homologous to that of the index case (97.1% homology in the overlapping sequence), even if the expressed sequence was longer (328 bp; Figure 2A). The insertion was located upstream of a different segment of retained intron at chr17:40333779, in which only the last 328 nucleotides were expressed in the fusion transcript, in-frame with RARA exon 3, and linked by 45 nucleotides of retained intron. Actually, the RARA intron 2 is 16.9 kb in length, and, in all PML/RARA-rearranged APL, the breakpoints are dispersed throughout the RARA intron 2 both within repetitive and unique regions.15 To the best of our knowledge, neither patient had clinical evidence of immune deficiency.
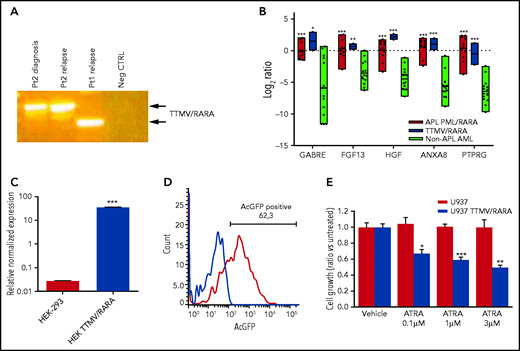
Phenotypic characterization of TTMV/RARA fusion transcript. (A) Detection of the TTMV/RARA chimeric fusion in cDNA of patient 2 diagnosis and relapse and of the index case for comparison. Patient 2 carried a longer insert sequence from TTMV ORF2, with respect to the index case (Pt1). (B) Overexpression of the top-scoring M3-AML–specific genes in the 2 cases carrying TTMV-RARA chimeric fusion (blue bars) similarly to the 11 APL cases from the SRA bioproject PRJNA721935 (red bars) and with respect to another 15 non-APL AML samples lacking cytogenetic and molecular aberrations (light green bars). The plot reports gene expression quantification as log2 ratios of transcripts per kilobase million (TPM); P value vs non-APL AML: *P < .05; **P < .01; ***P < .001. (C) Expression of TTMV-RARA by quantitative PCR in the HEK-293 cell line either parental or transfected with cloned full-length TTMV-RARA fusion transcript: P < .0001. (D) Flow cytometry assessment of TTMV/RARA fusion protein expression evaluated by means of AcGFP positivity in HEK-293 cells transfected with cloned full-length TTMV-RARA in frame with the AcGFP fluorescent tag sequence. (E) Effect of different ATRA concentrations on cell growth of U937 cells transfected with cloned full-length TTMV-RARA compared with the parental cell line after 4 days of treatment (*P < .05; **P < .01; ***P < .001).
Phenotypic characterization of TTMV/RARA fusion transcript. (A) Detection of the TTMV/RARA chimeric fusion in cDNA of patient 2 diagnosis and relapse and of the index case for comparison. Patient 2 carried a longer insert sequence from TTMV ORF2, with respect to the index case (Pt1). (B) Overexpression of the top-scoring M3-AML–specific genes in the 2 cases carrying TTMV-RARA chimeric fusion (blue bars) similarly to the 11 APL cases from the SRA bioproject PRJNA721935 (red bars) and with respect to another 15 non-APL AML samples lacking cytogenetic and molecular aberrations (light green bars). The plot reports gene expression quantification as log2 ratios of transcripts per kilobase million (TPM); P value vs non-APL AML: *P < .05; **P < .01; ***P < .001. (C) Expression of TTMV-RARA by quantitative PCR in the HEK-293 cell line either parental or transfected with cloned full-length TTMV-RARA fusion transcript: P < .0001. (D) Flow cytometry assessment of TTMV/RARA fusion protein expression evaluated by means of AcGFP positivity in HEK-293 cells transfected with cloned full-length TTMV-RARA in frame with the AcGFP fluorescent tag sequence. (E) Effect of different ATRA concentrations on cell growth of U937 cells transfected with cloned full-length TTMV-RARA compared with the parental cell line after 4 days of treatment (*P < .05; **P < .01; ***P < .001).
To ascertain that both patients harbored transcriptionally active RARA rearrangements, we analyzed the expression of the M3-oncogenic signature,16 showing that only the 2 cases displayed the overexpression of the top-scoring M3-specific genes, similarly to other pediatric APL and with respect to non-APL AML represented by normal-karyotype AMLs (Figure 2B). Last, to prove that the viral chimeric fusion was transcriptionally functional, we expressed the entire coding sequence of TTMV/RARA in frame with Aequorea coerulescens (AcGFP) fluorescent tag into HEK-293 cells, which resulted in a high expression of the chimeric transcript (Figure 2C) and in the presence of a significant population of AcGFP-positive cells (Figure 2D). Moreover, we showed that, besides increasing the percentage of CD11b+ cells, treatment with ATRA induced a significant decrease in cell growth of the U937 myeloid leukemia cell line transduced with TTMV/RARA compared with the parental cell line (Figure 2E), proving that TTMV viral insertion upstream of RARA exon 3 produces a functional chimeric protein.
Virus infections cause at least 12% of human cancers, but this is the first report suggesting a direct oncogenic role for an isolate of the Anelloviridae family. Anelloviruses are nonenveloped, circular, single-stranded DNA viruses,17 recently identified as one of the prevalent components of the human blood virome.18 Becausee Anelloviruses appear to be ubiquitous in the human population, are acquired very early in life, and maintain persistent active infections, they are thought of as nonpathogenic to human beings. However, increased viremia levels of Anelloviruses have been found in immune-suppressed individuals,19 whereas Anelloviridae viral load was linked to long-term nonprogression in HIV-infected children,20 and TTMV was associated with overall survival in pediatric patients undergoing lung transplant,21 suggesting that these viruses are normally kept under immunologic control. Nevertheless, this is the first report supporting a direct role of TTMV in malignant transformation, partly reinforced by the report of novel TTMV isolates in the plasma of patients with lymphoma or multiple myeloma.22,23
In summary, we herein report the first case of childhood APL carrying a TTMV/RARA chimeric transcript successfully treated with ATRA and ATO and subsequent HSCT, which supports the need to investigate the putative oncogenic role of TTMV in hematologic malignancies.
Acknowledgments
This work was partially supported by Associazione Italiana per la Ricerca sul Cancro grant MFAG2016, Id.19117 (R.M.) and University of Ferrara grant FIR-2020 (A.A).
Authorship
Contribution: A.A., D.M., S.N.B., and V.L. performed NGS and molecular analyses; A.A. and R.M. designed the project and supervised the teamwork; V.I., S.V., S.R., and M. Candela performed bioinformatic analyses; R.M., D.L., M. Carella, and A.P. collected clinical data; S.S. performed cytogenetic analyses; J.B. performed flow cytometry; A.P. supervised the study; and A.A., R.M., V.I., and A.P. analyzed data and wrote the paper, with contributions from all coauthors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Andrea Pession, Pediatric Unit, IRCCS Azienda Ospedaliero-Universitaria di Bologna, via Massarenti 11, 40138 Bologna, Italy; e-mail: andrea.pession@unibo.it.
Whole transcriptome sequencing data are available at the GEO repository with accession no. GSE169521.
Original data are available by e-mail request to the corresponding author.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal