

In this issue of Blood, 1 report that chromatin accessibility at regulatory regions of a heptad of transcriptional factors (TFs; LYL1, TAL1/SCL, LMO2, FLI1, ERG, GATA2, and RUNX1) displays distinct patterns at major stages of hematopoiesis and can thereby predict cell identity.
Hematopoiesis is strictly regulated by transcriptional networks, consisting of TFs, regulatory regions, and complexes of multiple transcriptional regulators interacting with each other.2 Regulatory regions are generally located in noncoding DNA sequences including intergenic and intragenic regions, and their epigenetic states such as DNA methylation and histone modifications dynamically control chromatin accessibility in a cell type–specific manner, and also contribute to cell fate decisions.3 Aberrant expression of key hematopoietic TFs or alterations in their regulatory regions, caused via genetic or nongenetic dysregulation, can rewire transcriptional networks and eventually lead to leukemogenesis.4 Although much work has been conducted to study the various effects of individual TFs or their regulatory regions on hematopoiesis and leukemogenesis, the role and function of larger transcriptional networks and their dynamics are far less well understood.
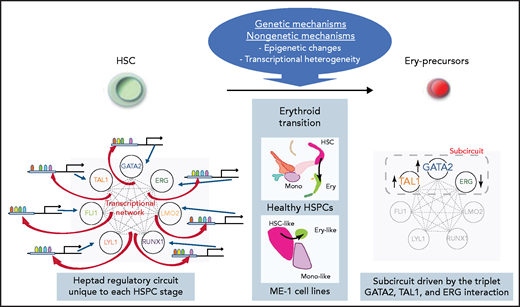
Schematic illustration of the transcriptional network consisting of heptad TFs and their regulatory regions and its dynamic change in erythroid transition.
Schematic illustration of the transcriptional network consisting of heptad TFs and their regulatory regions and its dynamic change in erythroid transition.
Prior work by Thoms and colleagues and other groups have demonstrated an important role and impact of a heptad of key TFs both in normal hematopoiesis and acute myeloid leukemia (AML). These factors shape a robustly interconnected circuit by controlling themselves and each other in human CD34+ human hematopoietic stem and progenitor cells (HSPCs), and play an essential role in stem maintenance.2,5 Furthermore, this heptad TF circuit was also reported to be active in AML, and heptad expression patterns were significantly associated with poor outcomes in patients.6 These earlier results clearly indicated that this TF heptad plays a vital role in HSPCs and AML cells, but one of the questions that remained unanswered was whether this circuit also plays a role in dynamic situations such as cell fate decisions during hematopoiesis and leukemogenesis. To this end, the present study set out to study dynamic changes of the TF heptad circuit at different stages of hematopoietic differentiation and the functional effect of this circuit on cell identity.
First, to explore the heptad circuit dynamics during normal hematopoiesis, Thoms et al analyzed single-cell RNA sequencing data of human HSPCs as well as chromatin accessibility data of sorted bone marrow cells. They found that the expression of heptad TFs and chromatin accessibility of their regulatory regions showed unique and different patterns throughout hematopoietic development, with a tendency for the heptad connectivity to disappear at the terminal differentiation stage. They then asked whether the accessibility of heptad regulatory regions was altered in AML cells at different stages including preleukemic hematopoietic stem cells (HSCs), leukemic stem cells, and blasts. Remarkably, chromatin accessibility patterns at each AML stage were distinct from one another, but again closely related to their biologically closest normal counterpart (eg, preleukemic HSCs compared with normal HSCs/multipotent progenitor cells), suggesting that the TF heptad circuit state is a defining feature of cell identity in both normal HSPCs and AML cells.
Thoms et al next investigated whether the connectivity of heptad TFs and their regulatory regions had a functional impact on heptad circuit activity. Chromatin immune precipitation sequencing data from human CD34+ HSPCs and 2 AML cell lines revealed a robust landscape of heptad TF patterns bound to their regulatory regions both in healthy and leukemic states, which were again quite similar with only minor differences. Furthermore, DNA-binding motif mutagenesis studies showed that the physical interaction of heptad TFs with their regulatory regions via consensus motifs were indeed essential for transcriptional activation (see figure). And finally, Thoms et al asked whether these regulatory circuits change dynamically during the shift from HSC to a precursor state. To address this, they performed semisupervised clustering of single-cell RNA sequencing data in ME-1 cells and uncovered an erythroid precursor-like subpopulation, which was characterized by high expression of TAL1 and GATA2, as well as low expression of ERG. In addition, they found that experimental overexpression of GATA2 or knock-down of ERG in ME-1 cells was able to induce transcriptional programs similar to the ones found in erythroid precursors. Combined, these findings support a model in that the triplet of GATA2, TAL1, and ERG shape a transcriptional subcircuit that is functionally relevant in HSC–erythroid transition.
Overall, the work from Thoms et al suggests that transcriptional networks dynamically control cell fate decisions and shape unique subcircuits during phenotype transition in healthy blood and leukemia. The study also raises several interesting questions for future study. The elegant work of the authors shows that the chromatin accessibility state of only a handful of enhancers can classify essentially all major stages of early hematopoiesis. They also succeeded in the identification of a unique transcriptional subcircuit during erythroid differentiation. Based on this, it will be interesting to identify and study other types of subcircuits which may govern the transition to precursors of other lineages. Another intriguing question is what precise mechanisms are harnessed to dynamically regulate the transcriptional networks in HSPCs, and which ultimately drive the transition from HSCs to precursors (eg, recent work has monitored low-level cofluctuations of PU.1, Gata1, and Gata2 in murine HSPCs) and found that transcriptional stochasticity of these TFs played a vital role in the maintenance of transcriptional plasticity.7 Future single-cell studies of changes of epigenetic states and transcriptional heterogeneity in the heptad circuit may lead to novel insights in that regard.
Another interesting question and future challenge are how the advances presented by Thoms et al could potentially be leveraged for therapeutic purposes. Although TFs are challenging drug targets in general, new approaches with small molecules or modified biologics are emerging.8-10 If future studies were able to identify AML-specific subcircuits which are not critical in normal hematopoiesis, the therapeutic targeting of such key TFs may be a highly promising strategy to tackle the generation of leukemic cell fates at its root.
Conflict-of-interest disclosure: U.S. has received research support and personal fees from Bayer Healthcare, personal fees from Celgene, research support from GlaxoSmithKline, research support and personal fees from Aileron Therapeutics, personal fees and involvement as scientific cofounder and member on the board of directors of Stelexis Therapeutics, personal fees from Pieris Pharmaceuticals, personal fees from Novartis, and personal fees from Vor Biopharma outside the submitted work. G.T. declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal