

Formation of neutrophil extracellular traps (NETs) during fetal development and in neonates is suppressed by circulating NET-inhibitory peptides (NIPs). In this issue of Blood, show that 1 NIP is formed by serine protease high temperature requirement A1 (HtrA1) cleavage of α-1 antitrypsin (A1AT).1
Neutrophils are innate immune cells that defend against infectious microbes, including bacteria, fungi, viruses, and parasites.2 Neutrophils use 3 major strategies to eliminate pathogens: phagocytosis, degranulation, and NETs. NETs are web-like chromatin structures studded with cytosolic proteins and granule enzymes that are extruded from neutrophils to capture, immobilize, and kill microbes. Although NETs are an effective host defense against infections, dysfunctional NET formation causes tissue damage and is thought to be involved in the pathology of many diseases, including autoimmune disorders, diabetes, atherosclerosis, cancer, sepsis, and preeclampsia (PE).2,3 If excess NET formation is harmful, do we have any mechanisms to protect our body from the dark side of NETs? In this issue, Campbell et al answer this interesting question using a model of neonatal sepsis.
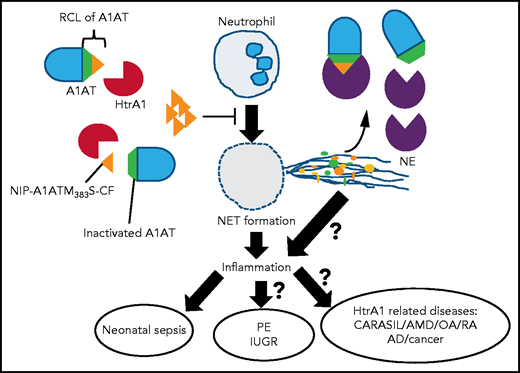
HtrA1 secreted from fetal syncytiotrophoblasts cleaves A1AT and generates NIP–A1ATM383S-CF in fetal blood vessels. NIP–A1ATM383S-CF inhibits excess NET formation in neonates just after birth and ameliorates neonatal sepsis. A1AT cleaved by HtrA1 (in the middle of RCL) loses its inhibitory activity against NE. The balance of these 2 opposite mechanisms might regulate inflammation during neonatal sepsis. It is an important issue whether HtrA1 inhibits NET formation in the intervillous space through NIP–A1ATM383S-CF and, thereby, is involved in the pathogenesis of PE and IUGR. It is possible that similar mechanisms underlie other HtrA1-related diseases, such as cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL), age-related macular degeneration (AMD), osteoarthritis (OA), rheumatoid arthritis (RA), Alzheimer disease (AD), and cancer.
HtrA1 secreted from fetal syncytiotrophoblasts cleaves A1AT and generates NIP–A1ATM383S-CF in fetal blood vessels. NIP–A1ATM383S-CF inhibits excess NET formation in neonates just after birth and ameliorates neonatal sepsis. A1AT cleaved by HtrA1 (in the middle of RCL) loses its inhibitory activity against NE. The balance of these 2 opposite mechanisms might regulate inflammation during neonatal sepsis. It is an important issue whether HtrA1 inhibits NET formation in the intervillous space through NIP–A1ATM383S-CF and, thereby, is involved in the pathogenesis of PE and IUGR. It is possible that similar mechanisms underlie other HtrA1-related diseases, such as cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL), age-related macular degeneration (AMD), osteoarthritis (OA), rheumatoid arthritis (RA), Alzheimer disease (AD), and cancer.
The same research group previously discovered that fetal and neonatal neutrophils from umbilical cord blood failed to form NETs as a result of endogenous neonatal NET inhibitory factors.4 They found that one was the C terminus of A1AT.5 They suspected that HtrA1 was the protease responsible for the cleavage of A1AT for the following reasons. First, because the NET-inhibitory activity was higher in umbilical cord blood plasma than in adult plasma, they believed that placental proteases cleaved A1AT to generate NIPs.5 Second, it was reported recently that placentally expressed protease HtrA1 cleaved A1AT after Met-383, which generates C-terminus fragments of A1AT that are very similar in size and amino acid composition to NIPs.6
Campbell et al detected robust HTRA1 expression in fetal syncytiotrophoblast cells of the human placenta, as previously reported. HTRA1 expression levels in term and preterm umbilical cord plasma was significantly higher compared with the plasma of healthy adults. However, HTRA1 levels in neonatal plasma decreased rapidly to levels similar to those of healthy adults 3 days after delivery. Along with reduced HtrA1 levels in neonatal plasma, 4-kDa NIP disappeared from neonatal plasma 3 days after delivery. The decrease in HtrA1 and NIP in neonatal plasma after birth suggested that the placenta was the source of HtrA1. The investigators confirmed that HtrA1 cleaved A1AT in a cell-free system to generate the 4-kDa peptide A1ATM383S-CF and verified that A1ATM383S-CF was identical to NIP by mass spectrometry. Synthetic A1ATM383S-CF inhibits NET formation stimulated by phorbol-12-myristate acetate or lipopolysaccharide in a dose-dependent manner in human neutrophils in vitro. A1ATM383S-CF inhibited nuclear condensation, which is an initial step in NETosis, a special type of cell death leading to NET formation. However, A1ATM383S-CF failed to directly inhibit neutrophil elastase (NE) activity.
To extend their in vitro findings using human samples to an in vivo mouse model, they evaluated NET formation in pup plasma of wild-type (WT) mice. They confirmed that NET-inhibitory activity in pup plasma immediately after birth was also conserved in mice, which was gradually reduced from days 7 to 10 after birth. NET-inhibitory activity in the pup plasma of WT mice was exactly correlated with the presence of NIPs. HtrA1 and NIP were significantly decreased in HTRA1−/− pups. It is important to note that they showed for the first time that the neonatal NET inhibition system is conserved in humans and mice. Finally, in a preclinical model of neonatal sepsis, inhibition of NETs by treatments with A1ATM383S-CF increased survival of WT and HTRA1−/− pups, which was unexpectedly higher in HTRA1−/− pups than in WT pups. From this study, it seems that excess NET formation actually worsens neonatal sepsis. A1AT, a member of the serine protease inhibitors superfamily (SERPINs), is an important inhibitor of NE. The cleavage of A1AT by HtrA1 after Met-382, which is in the middle of the reactive center loop (RCL; aa 368-392) of A1AT, results in a loss of its inhibitory function against NE.6 It is an interesting possibility that HtrA1 regulates inflammation by balancing 2 opposing functions: downregulation by inhibiting NETs and upregulation by activating NE (see figure). This complicated mode of regulation of neutrophil functions by HtrA1 may explain why both elevations and reductions in HtrA1 levels cause a variety of human diseases.7
NET formation is detected in the intervillous space, a maternal-fetal interspace, even in placentas of normal pregnancies; however, it is dramatically increased in the placentas of pregnancies with PE.3 Dysregulated HtrA1 expression in placenta and in maternal plasma has repeatedly been reported in abnormal pregnancies, for example, PE and intrauterine growth restriction (IUGR).8 One of the subsequent important issues is whether placental HtrA1 inhibits dysregulated excess NET formation to maintain immune tolerance at the maternal-fetal interface and prevents abnormal placentation and gestation leading to PE-IUGR (see figure).
HtrA1 is a member of HtrA family proteins that are distributed in a range of species from bacteria, yeast, and plants to humans.7,9 Bacterial HtrAs are heat shock proteins that degrade or refold proteins that are denatured or unfolded under different stress conditions. Mammalian HtrA1 expression is also induced by several stresses, including oxidative stress and endoplasmic reticulum stress. In humans, loss-of-function mutation of the HtrA1 gene is the cause of the hereditary small vessel disease, cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy.10 Furthermore, abnormal expression and activity of HtrA1 are related to various inflammatory diseases, such as age-related macular degeneration, rheumatoid arthritis, osteoarthritis, Alzheimer’s disease, and cancer.7 Another important issue is if and how NET formation regulated by HtrA1 is involved in the pathogenesis of these HtrA1-related diseases (see figure). Certainly, this article will spur additional work on the regulation of NETs.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal