


Abstract
Hematological malignancies are an aggregate of diverse populations of cells that arise following a complex process of clonal evolution and selection. Recent approaches have facilitated the study of clonal populations and their evolution over time across multiple phenotypic cell populations. In this review, we present current concepts on the role of clonal evolution in leukemic initiation, disease progression, and relapse. We highlight recent advances and unanswered questions about the contribution of the hematopoietic stem cell population to these processes.
Introduction
Leukemias, like many tumors, contain diverse populations of cellular clones. This tumor heterogeneity is further shaped by clonal evolution, a process by which accumulated aberrations, both genetic and nongenetic, generate diversity that is subject to selective pressures. The clonal makeup changes during disease pathogenesis, whether it be initial malignant transformation, disease progression, or relapse. Historically, clonal evolution has been thought of as a primarily linear process in which a series of subsequent mutations give rise to novel subclones that drive disease progression.1 Recently, many lines of evidence point to a highly dynamic process of clonal evolution in leukemia that originates long before diagnosis, including at the stem cell level, and a clonal landscape that is continuously reshaped in response to changing selective pressures. Having a greater understanding of clonal evolution and its origins is of particular importance in myelodysplastic syndromes (MDSs) and acute myeloid leukemia (AML), where poor clinical outcomes are common, in large part because of high rates of progression and relapse even after complete remission.2-4 This review provides an overview of emerging concepts of clonal evolution, specifically in the context of myeloid malignancies and highlights recent work proposing premalignant stem cells as a clinically relevant source of clonal heterogeneity.
Clonal evolution in leukemic initiation
Over time, competition between hematopoietic stem cells (HSCs) drives clonal diversification of the hematopoietic system. In some cases, a series of transforming events give rise to preleukemic stem cells (pre-LSCs), which precedes the transformation of leukemia stem cells (LSCs) from which the tumor bulk is derived.5,6 Early evidence for this concept was found in murine models of preleukemic-to-overt leukemia transition,7-9 followed by the discovery of transcriptional and other epigenetic changes in phenotypically “normal” stem cells from patients8,10-13 and, later, genomic identification of pre-LSCs in patients with AML6,14,15 and MDS.16
Myeloid malignancies are often preceded by a premalignant state in which accumulated cellular alterations can exist years prior to the development of symptoms. In some cases, the earliest events arise as germline mutations, as seen in familial leukemias.17 Often, somatic mutations resulting in clonal hematopoiesis (CH) can precede transformation.18-23 Although most cases of CH do not progress to malignancy, recent studies directly linking CH mutations to AML suggest that AML can be descended from long-lived premalignant CH clones and provides additional evidence supporting the stem cell origin of leukemias.23-25
The effects of clonal selection during initiation can be exemplified by therapy-related MDS and AML. Patients who have undergone previous chemotherapy for unrelated malignancies demonstrate a higher rate of CH, especially in genes related to DNA damage response, such as TP53 and PPM1D.26,27 These mutations suggest that genotoxic stress caused by therapy can lead to the expansion of DNA damage-resistant clones. This is further supported by the enrichment of these mutations in therapy-related MDS and AML compared with de novo MDS/AML, without an increase in the absolute number of mutations.28,29
Collectively, these findings, combined with the longevity and long-term self-renewal capacity of stem cells,30,31 suggest that the HSC population naturally diversifies over time. Continued accumulation of aberrations leads to the transformation of pre-LSCs and the appearance of a clinically detectable tumor bulk, or blast cells. Although the majority of blast cells usually belong to a limited number of dominant clones, the processes of evolution, competition, and selection during premalignancy and transformation result in a complex and heterogeneous clonal architecture, often including numerous minor subclones. This concept has been reflected in analysis of the clonal makeup of MDS and AML bulk cell populations by single-cell or deep next-generation sequencing, revealing diverse clones with patterns of linear and branching evolution.32-40
Although mutations provide an easy way to track clones and their evolution, nongenetic factors also play an important role in clonal selection and disease initiation.41-44 For example, transcriptional and other RNA regulatory changes appear to be key events in leukemic transformation, with genetic mutations being only a part of that dysregulation.18,45-51 Additionally, cell-extrinsic mediators can affect how HSCs respond to cell-intrinsic aberrations.52-55 These effectors can include extrinsic selective pressures, such as chemotherapy, as well as a chronic inflammatory milieu and decreased supportive function of an aging bone marrow niche.56,57 A deteriorating hematopoietic microenvironment may promote the preferential expansion of certain mutated clones that would have had no advantage in an unperturbed system. To that end, it is unclear whether CH is necessarily a pathological step on the path to leukemogenesis or simply a result of natural clonal diversification in response to a changing environment. This idea is also supported by the persistence of some common CH mutations after complete remission without an increased risk for relapse.58
Clonal evolution in progression and relapse
Clonal evolution continues as part of the natural history of the disease, but the evolutionary patterns and role of the stem cell compartment become more difficult to track at disease stages when a large dominant bulk population is present. Cytotoxic and targeted treatments can impose major selective pressures, overpowering cell-intrinsic advantages to direct clonal evolution. Paired MDS and AML sequencing studies have generally shown persistence of the dominant mutations and karyotype in progressive disease, often with the emergence of a newly detected subclone.32-36 For example, treatment with lenalidomide showed successful suppression of the del(5q) clone in MDS; however, in most cases treatment was associated with selection and expansion of a preexisting minor subclone.32,33 This suggests that relapse following treatment may often be a result of clonal selection of an existing clone and not induced mutagenesis. Although, in many cases, the dominant clone after treatment contained most or all mutations found in the dominant clone at diagnosis, in some cases a distinct mutational profile with few common mutations was detected, indicating early divergence from the dominant lineage at diagnosis.34,36 Conceptually similar phenomena have also been observed in myeloproliferative neoplasms.59-62
In AML, early studies of paired diagnosis-relapse samples demonstrated that most cases of AML with chromosomal abnormalities remained cytogenetically stable, sometimes acquiring additional chromosomal abnormalities.63 However, there were some cases in which relapse clones were cytogenetically distinct. Genomic analysis of paired samples further established that AML relapse can be due to evolution, often linear, of the dominant disease clone. In other cases, AML relapse originated from a related, but distinct, clone.15,37 In either of these cases, the relapse clones have sometimes been detectable in primary disease as a minor clone.38,64,65
These studies and others suggest that preexisting minor clones may be critical to disease progression, whether they are directly derived from the dominant clone or not. Even in cases in which the minor clones are not detected, they may be present. Classical methodologies of investigating the tumor bulk make it difficult to distinguish whether a newly detected clone at progression is an expansion of an existing leukemic clone below detection limits in primary disease or genuinely due to mutational acquisition since prior sampling. Distinguishing between these important, but not necessarily mutually exclusive, possibilities will have obvious implications for strategies of targeted diagnostics and precision therapeutics to achieve lasting disease control or even prevention of relapse and progression.
The stem compartment in progression and relapse
The stem cell compartment plays an important role in the pathogenesis of myeloid malignancies, not only as the source of the cancer, but also because LSCs and pre-LSCs persist throughout the course of the disease. LSC burden has been shown to be an excellent predictor of clinical outcomes in AML,66 and LSCs are thought to cause relapse when not fully eradicated.67,68 HSCs are also subject to various cell-extrinsic selective pressures, such as niche effects, inflammation, and immunological control, which can codrive clonal evolution.42,54,69-72 However, their role during disease progression, including in the context of therapeutic interventions, has not been fully appreciated until recently.
The origin of a clone can be traced back by using specific somatically acquired genetic variants as lineage-tracing markers. Cell populations that share the same variants can be assumed to originate from the same founding cell, irrespective of whether the specific variant is assumed to confer altered functionality.73 To determine the origin of secondary disease, Shlush et al,74 Chen et al,16 and Quek et al75 applied this approach to directly investigate the role of stem cell populations in AML relapse, MDS to secondary AML (sAML) progression, and targeted therapy of AML, respectively.
Shlush et al determined the mutational profile of bulk AML samples at diagnosis and relapse in 11 patients, as well as sequencing nonleukemic T cells to distinguish between leukemic and preleukemic mutations. Most patients had a large number of unique relapse variants not detected in the diagnosis sample, consistent with a clonal switch at relapse. To characterize the genetic diversity of the stem cell population, they performed xenotransplantation of the diagnosis sample and assayed the resultant xenografts for the presence of relapse variants. In addition, sorted progenitor populations from the primary sample were assayed when available. Variant allele frequencies were then used to infer the clonal evolution from diagnosis to relapse. This analysis demonstrated that most relapse variants were already present at the time of diagnosis to varying degrees, suggesting that chemotherapy-induced mutations were not the cause of relapse; instead, the stem cells destined to give rise to relapse clones were preexisting and resistant to therapy. Interestingly, relapse variants were never detected in primary T-cell populations or in lymphoid xenograft populations, suggesting LSCs as the most primitive probable source of these clones, and not pre-LSCs, in relapsed AML. Shlush et al identified 2 major patterns of LSC source populations responsible for relapse: relapse origin‐primitive and relapse origin‐committed. In the relapse origin‐primitive group, mutations found only in the relapse blasts, but not in the diagnosis blasts, were detectable in early progenitors, as well as in the diagnosis xenografts. In these patients, the LSCs responsible for relapse are exceedingly rare at diagnosis and seemingly do not contribute to the blasts at that time. However, they are apparently resistant to standard treatment and instead are positively selected and reinitiate the leukemia during relapse. In the relapse origin‐committed group, the relapse clones are more closely related to the major diagnosis clone present and originate from more committed progenitors; however, they still possess a strong stemness signature.
In their study of MDS progression, Chen et al assayed longitudinal paired samples from 7 patients with high-risk MDS who had progressed to sAML to interrogate the clonal architecture of the stem and blast populations. Phenotypically defined and functionally validated pre–MDS-stem cells (pre–MDS-SCs), MDS-SCs, pre-LSCs, LSCs, and blast populations were sorted from bone marrow samples at both time points. The mutational profiles of these samples were determined by targeted deep sequencing, followed by single-cell validation. The stem populations showed greater mutational diversity and an increased number of mutational clusters (ie, clones) than did the blast population. This further demonstrates that the stem population harbors increased heterogeneity that is not appreciated in mature populations.
By tracking the variant allele frequencies in each population, Chen et al were able to infer the clonal evolution that occurs during MDS to sAML progression. Most patients (6/7) showed a demonstrable clonal shift in the blast populations upon progression. However, all patients showed persistence of ≥1 dominant clone after transition, indicating that the bulk of sAML cells likely originated from the same founding population, consistent with previous results. In 4 patients, mutations in a dominant clone that seemed to newly emerge in the AML blasts (ie, was not detectable in the MDS blasts) were detectable in the MDS-SC or pre–MDS-SC population at the MDS time point. In 1 of these cases, the clone was highly represented (49%-52% clonal prevalence) in the MDS stem populations, pointing to possibly complex differential competition and selection mechanisms of stem cell subclones at the MDS and sAML disease stages, respectively, that may not be predicted based on clone size alone. Similar results, of seemingly new sAML mutations being detected in the stem cell compartment at the MDS stage, were reported by Simonsen et al76 in 2 of 3 paired MDS-sAML samples assayed.
In their study, Quek et al detailed the clonal basis of response and acquired resistance to enasidenib. In patients with IDH2-mutant AML, treatment with enasidenib produces a clinical response by promoting differentiation of arrested leukemic cells. By sorting multiple stem and progenitor populations of longitudinal samples, Quek et al tracked clonal changes across different cell populations and in response to treatment. In some cases, the clonal compositions differed across cell populations, especially when comparing more committed cells to early stem and progenitor cells. In most responding patients, the terminally differentiated mature cells arose from the dominant AML clone, retaining the same mutational profile. In other cases, treatment induced differentiation and selection of ancestral or seemingly unmutated clones. Relapse and reimposition of the differentiation block were found to be a result of clonal evolution or selection. In 1 patient with 2 divergent dominant clones (both IDH2 mutant) present at diagnosis, treatment induced a response in only 1 of the clones. At relapse, the nonresponding clone shrunk in most populations and was only found with high prevalence in lymphoid-primed multipotent progenitors. However, accumulation of additional mutations in the persistent nonresponding clone led to selection of that clone and relapse. These findings highlight the complexities of clonal evolution that can take place during AML disease course and targeted therapy and show the importance of studying stem and progenitor populations directly in the clinical context.
These findings point to a model in which some clones diverge from the dominant leukemic lineage very early in disease pathogenesis and including at the level of premalignant stem cell subclones. These coexisting stem cell clones continue to evolve in parallel with the clinically apparent dominant clone and have the capacity to be selected for at a later point (Figure 1).
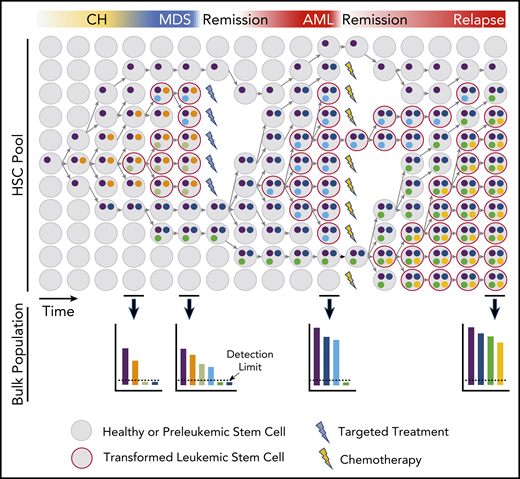
Model of HSC clonal evolution leading to MDS and AML pathogenesis and progression. Schematic depiction of a hypothetical evolutionary history in the stem compartment and its representation in bulk samples. Clonal evolution begins with the acquisition of mutations or other nongenetic aberrations that result in clonal expansion and further mutation of the founder clone. A descendent HSC may gain a new aberration, causing it to branch into a new subclone. Branching subclones continue to evolve in parallel and may have disparate responses to selective pressures. Some subclones expand rapidly upon acquisition of a new mutation, whereas others may remain stable for longer periods. Extrinsic pressures, such as targeted therapies, may affect only some clones, whereas nontargeted therapies may have a more widespread, but often also less deep, effect. Clonal outgrowth is represented with time on the x-axis and relative abundance of HSC clones on the y-axis. The colors represent accumulated aberrations. The lower panels show the relative frequency of each alteration found in the bulk blood population at selected time points. Bulk sampling may not reflect the complete diversity and complexity found in HSCs, because variants below the detection limit (dotted line) will not be accounted for but may be shown to be disease relevant at later time points.
Model of HSC clonal evolution leading to MDS and AML pathogenesis and progression. Schematic depiction of a hypothetical evolutionary history in the stem compartment and its representation in bulk samples. Clonal evolution begins with the acquisition of mutations or other nongenetic aberrations that result in clonal expansion and further mutation of the founder clone. A descendent HSC may gain a new aberration, causing it to branch into a new subclone. Branching subclones continue to evolve in parallel and may have disparate responses to selective pressures. Some subclones expand rapidly upon acquisition of a new mutation, whereas others may remain stable for longer periods. Extrinsic pressures, such as targeted therapies, may affect only some clones, whereas nontargeted therapies may have a more widespread, but often also less deep, effect. Clonal outgrowth is represented with time on the x-axis and relative abundance of HSC clones on the y-axis. The colors represent accumulated aberrations. The lower panels show the relative frequency of each alteration found in the bulk blood population at selected time points. Bulk sampling may not reflect the complete diversity and complexity found in HSCs, because variants below the detection limit (dotted line) will not be accounted for but may be shown to be disease relevant at later time points.
Conclusions
With the current ease of DNA sequencing and the implementation of newer methods with increasingly higher cellular resolution, it has become clear that clonal heterogeneity is present in all hematological malignancies,77-80 including at the stem cell level.16,61,62,74,75 The clonal architecture found in the tumor bulk is a result of the continuous accumulation and selection of genetic and nongenetic variants, first in HSCs and later in the cancer cells themselves. These recent findings highlight the stem cell compartment as an additional space where clinically relevant clonal evolution is occurring during the disease process.
The evolutionary pattern observed in a given case after treatment likely depends on the treatment’s efficacy. If the dominant clone persists after treatment, the secondary clone will probably show a linear evolutionary pattern with greater genetic similarity to the primary clone. New targeted therapies, such as IDH and FLT3 inhibitors, or other emerging therapies resulting in broader and deeper responses, such as venetoclax plus azacitidine, will likely be more successful in ablating the initial dominant clone and may result in relapse clones that are more divergent. Ultimately, treatments that target the tumor evolutionary process will likely need to be considered to achieve more effective and lasting disease control. Such approaches could include treatments directly targeting phenotypic pre-LSCs70 or specific vulnerabilities of clones carrying CH mutations81 or treatments affecting mutational acquisition and subclonal diversification.82-84 The targeting of the inflammatory environment that drives selection is also emerging as an interesting new avenue.54,85-90 Furthermore, the direct targeting of transcriptional regulators, previously deemed “undruggable,” is becoming increasingly feasible through chemical and biophysical advances and the design and optimization of novel therapeutic substance classes,82,91-94 some of which have recently advanced to the clinic. Lastly, the ability to detect relevant minor subclones and understand what drives the expansion of a seemingly inactive pre-LSC clone may lead to additional anticipation-based treatment strategies.
However, there is much left unknown, especially at the stem cell level. The degree of heterogeneity is almost certainly still underestimated as a result of technical limitations. There are likely hundreds, and possibly more, stem cell subclones that exist and contribute to the fitness of the HSC pool, and it may be difficult to determine which are clinically relevant. Deep-sequencing or single-cell analysis of multiple longitudinal stem cell and blast samples collected during disease course and treatment could help to determine the variants that contribute to cellular fitness and subsequent expansion and in relation to the specific treatment context. Because of the observed long-lived presence of minor stem cell subclones that do not obtain dominance, despite well-known genetic driver mutations, the mechanisms are unlikely to be solely genetic and may include cell-extrinsic mechanisms. Therefore, comprehensive functional studies, with human (pre)cancer stem cells, but also newly emerging polyclonal (and nonlinear) models of leukemia pathogenesis (eg, in the mouse, zebrafish, or others), are warranted to better understand the factors governing fitness of the heterogeneous stem cell pool and, ultimately, how it can be influenced pharmacologically, including in a preemptive manner.
Acknowledgments
The authors thank Emily Schwenger and Amit Verma for helpful comments and critical discussion of data and ideas discussed in this article. They apologize to all scientists whose work could not be cited because of space limitations in this spotlight format.
J.S. is supported by National Institutes of Health (NIH)/National Institute of General Medical Sciences grant T32GM007288. U.S. is supported by NIH/National Cancer Institute grant R01CA217092, the Albert Einstein Cancer Center core support grant P30CA013330, and is the Diane and Arthur B. Belfer Faculty Scholar in Cancer Research of the Albert Einstein College of Medicine.
Authorship
Contribution: J.S., J.M.G., and U.S. wrote the manuscript.
Conflict-of-interest disclosure: U.S. has received research funding from GlaxoSmithKline, Bayer Healthcare, Aileron Therapeutics, and Novartis; has received compensation for consultancy services and for serving on scientific advisory boards from Novartis, GlaxoSmithKline, Bayer Healthcare, Celgene, Aileron Therapeutics, Stelexis Therapeutics, and Pieris Pharmaceuticals; and has equity ownership in and serves on the board of directors of Stelexis Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Ulrich Steidl, Albert Einstein College of Medicine, 1300 Morris Park Ave, Chanin Bldg, Rm 601-605, The Bronx, NY 10461; e-mail: ulrich.steidl@einsteinmed.org.
