


Abstract
Burkitt lymphoma (BL) is a highly aggressive, B-cell, non-Hodgkin lymphoma categorized into endemic, sporadic, and immunodeficiency-associated subtypes. BL has distinct pathologic and clinical features, characterized by rapidly progressive tumors with high rates of extranodal involvement. Next-generation-sequencing analyses have further characterized the genomic landscape of BL and our understanding of disease pathogenesis, although these findings have yet to influence treatment. Although most patients are cured with intensive combination chemotherapy, given the paucity of randomized trials, optimal therapy has not been defined. Furthermore, treatment of elderly patients, patients with central nervous system involvement, or those with relapsed disease remains an unmet need. In this review, we highlight the clinical, pathologic, and genomic features, as well as standard and emerging treatment options for adult patients with BL.
Introduction
Burkitt lymphoma (BL) is a highly aggressive, B-cell, non-Hodgkin lymphoma (NHL) characterized by the translocation and dysregulation of the protooncogene MYC. Although initial reports of BL date back to the early 20th century, BL derives its name from the surgeon Denis Burkitt, who in the 1950s, described cases of rapidly progressive, unusual jaw tumors in children in Uganda, which were later identified as lymphoma.1,2 Since these initial descriptions, it has been recognized that BL can occur outside of Africa in both pediatric and adult populations. The World Health Organization now recognizes 3 distinct subtypes of BL, endemic, sporadic, and immunodeficiency-associated disease.3
Clinical features of BL subtypes
Endemic BL is highly prevalent in equatorial Africa and represents the most common pediatric malignancy in sub-Saharan Africa.4 Endemic BL occurs with a 2:1 male predominance and at a median age of 6 years.5 This variant is universally associated with Epstein-Barr virus (EBV), suggesting a direct causative role of the virus in lymphoma pathogenesis. Endemic BL is also largely restricted to geographic regions in which Plasmodium falciparum malaria is holoendemic. It has been proposed that chronic B-cell activation or promotion of the oncogenic potential of EBV in the setting of malaria coinfection promotes oncogenesis.6-8 Patients typically present with rapidly enlarging masses of the jaw or periorbital region. Although involvement of the bone marrow is not commonly seen at diagnosis, other extranodal sites, including the gastrointestinal tract, adrenals, kidneys, and gonads, are commonly involved.
Sporadic BL occurs worldwide and represents most cases occurring in the United States and Western Europe. Sporadic BL is more commonly seen in pediatric patients, where it represents 20% to 30% of lymphomas. In adults, sporadic BL rarely occurs, comprising ∼1% of cases of NHL in the United States.9 Sporadic BL also occurs more commonly in males, with more than a twofold increase in men as compared with women.9,10 In contrast to endemic BL, EBV association is less prevalent.11 Sporadic BL frequently involves extranodal sites, particularly the central nervous system (CNS), which is often leptomeningeal rather than parenchymal, gastrointestinal tract, and bone marrow.9 In a recent retrospective study of BL in adults in the United States, 19% of patients had CNS involvement at the time of diagnosis, 16% of which was leptomeningeal disease.12
The third subtype of BL occurs in the setting of immunodeficiency, most commonly, HIV. This variant comprises ∼20% of the cases of BL in the United States.5 Of note, as BL typically occurs with relatively well-preserved CD4 counts, the prevalence has not significantly changed in HIV patients with the advent of highly active antiretroviral therapy.13 Immunodeficiency-associated BL typically presents with nodal involvement, but additional sites, including the bone marrow and CNS, can be involved. When adjusted for prognostic factors, patients with HIV-associated BL can have similar outcomes as compared with those with HIV− disease.14
Pathology
BL is characterized histologically by complete effacement of the lymph node architecture by sheets of lymphocytes. The tumor cells are typically intermediate in size and nonpleomorphic and contain basophilic cytoplasm, prominent vacuoles, and round nuclei (Figure 1).15 The Ki-67, a measure of growth fraction, typically approaches 100%. Abundant large and irregularly shaped macrophages, which have ingested apoptotic tumor cells, are interspersed among the lymphocytes to give the classic “starry-sky” appearance (Figure 1).
![Representative pathologic specimens of BL. (A) Hematoxylin and eosin slide demonstrating abundant large and irregularly shaped macrophages interspersed among lymphocytes to give the classic “starry sky” appearance. (B) BL cells with prominent cytoplasmic vacuoles. Reproduced from the ASH Image Bank (images 00061346 [A] and 00001117 [B]) with permission.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/6/10.1182_blood.2019004099/3/m_bloodbld2019004099cf1.png?Expires=1769079715&Signature=gnVIQ3WJAs8XpX~n5IZyovppt6~XiVpMlFHRdJQFNDUNVpdrUeNsvtaLFryD-0z4MOyEMtzPIzlA-K46cuFZH~GnaRDisjghkWfIWU-W9JMXlQ9Wp4R2cnmd8iJ9xkS6U7NPbfpFOgP8XJJDiqmGHo-43nahek3ROybIPTPsJFoepc0CsVp2Eh7Ho4niw7vCfyl4XvbrMyOAVqddnwryKPVxN6RHOcvxpIEkYBzx~~nYcZguSVeQ-gWiXbRt1gXwRhdSY20kkM5y77qF2wYgicDR70PHOV9mnhS3xzDpRV3~~UbzGTzqfZB-zJhrBbXvBA5lhaeJG9vOt~6zc1vuBQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Representative pathologic specimens of BL. (A) Hematoxylin and eosin slide demonstrating abundant large and irregularly shaped macrophages interspersed among lymphocytes to give the classic “starry sky” appearance. (B) BL cells with prominent cytoplasmic vacuoles. Reproduced from the ASH Image Bank (images 00061346 [A] and 00001117 [B]) with permission.
Representative pathologic specimens of BL. (A) Hematoxylin and eosin slide demonstrating abundant large and irregularly shaped macrophages interspersed among lymphocytes to give the classic “starry sky” appearance. (B) BL cells with prominent cytoplasmic vacuoles. Reproduced from the ASH Image Bank (images 00061346 [A] and 00001117 [B]) with permission.
BL cells are thought to be derived from the germinal center or postgerminal center of the lymph node, as they typically contain mutations in the variable region of the immunoglobulin genes, a hallmark of germinal center transit. By immunohistochemistry, malignant cells are positive for CD20, CD10, BCL6, CD79a, HLA-DR, and CD45 and are negative for CD5, BCL2, TdT, and CD23.16,17 EBV expression is uniformly seen in endemic BL and detected in 25% to 40% of sporadic and immunodeficiency-associated cases.15
Of note, tumors resembling BL histologically without MYC translocations have been described, often with concurrent 11q alterations. Compared with BL, these lymphomas often have more complex karyotypes and lower levels of MYC expression and frequently present with nodal involvement.3 Despite otherwise histologic and clinical similarities, these lymphomas have ultimately been classified as a separate entity by the World Health Organization.3 BL must similarly be distinguished from other high-grade NHLs, including high-grade B-cell lymphoma not otherwise specified, high-grade B-cell lymphoma with concurrent MYC and BCL-2 and/or BCL-6 translocations, and diffuse large B-cell lymphomas (DLBCLs) with MYC translocations, which can have overlapping histologic features and cytogenetic aberrations. Although DLBCLs are characterized by a pan–B-cell marker, such as CD20, CD19, CD22, and CD79a that can overlap with BL, they can also express BCL-2, and rarely CD30 and CD5, which are less likely seen with BL.17 Histologically, DLBCLs also typically appear more heterogenous with larger cells with prominent nucleoli and abundant cytoplasm, rather than the intermediate-sized and uniform cells that create the starry-sky appearance that is seen with BL.17
Genetics
BL was the first lymphoid malignancy in which a chromosomal translocation was linked to disease pathogenesis.18,19 The defining genetic hallmark is the reciprocal translocation of the MYC gene, located on chromosome 8, to the immunoglobulin heavy chain (IGH) locus on chromosome 14 (t(8;14)). More rarely, translocations of MYC can involve either the κ light chain on chromosome 2 [t(2;8)] or the λ light chain on chromosome 22 [t(8;22)]. The translocation of MYC with either the immunoglobulin heavy or the light chain locus results in constituent activation and overexpression of MYC, which serves as an oncogene to promote growth and proliferation (Figure 2). In BL, malignant cells are of a centroblast phenotype, which are normally characterized by rapid proliferation and somatic hypermutation, as well as MYC repression by BCL-6.20 Thus, in BL, MYC is able to activate a host of genes that augments the centroblast phenotype, allowing for acquisition of additional cooperative alterations.21,22 Acquisition of mutations impairing TP53, for example, which occurs in up to 35% of BL, further impairs apoptosis.22
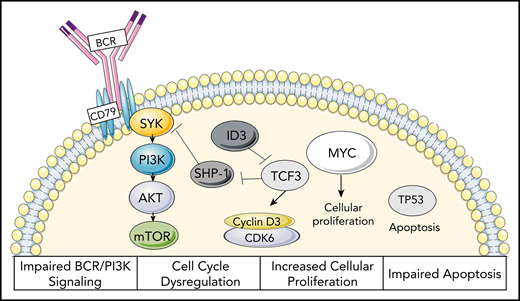
Proposed oncogenic mechanisms of BL. Characterization of impaired BCR/PI3K signaling, cell-cycle dysregulation, increased cellular proliferation, and impaired apoptosis seen in BL.22
Proposed oncogenic mechanisms of BL. Characterization of impaired BCR/PI3K signaling, cell-cycle dysregulation, increased cellular proliferation, and impaired apoptosis seen in BL.22
Initial studies utilizing gene expression profiling identified a distinct genetic profile in BL characterized by high expression of MYC target genes and a subgroup of germinal-center B-cell genes and decreased expression of major histocompatibility complex class I genes and NF-κB targets, allowing for improved differentiation of BL from DLBCLs.23,24 Subsequently, integrated genome, exome, and transcriptome analyses were used to identify recurrent mutations in BL. In these studies, the transcription factor TCF3 or its negative regulator ID3 was found to be mutated in ∼70% of sporadic and immunodeficiency-associated BL and 40% of endemic cases.3,25-27 TCF3 is one of the main regulators of the centroblast program that promotes survival and proliferation through activation of B-cell receptor (BCR)/phosphatidylinositol 3-kinase signaling pathways. The tonic BCR signaling in BL differs from that seen in DLBCLs, as it does not result in increased expression of NF-κB target genes.28 Mutations were also found in cyclin D3, occurring in 30% of BL, resulting in cell-cycle dysregulation (Figure 2).3
More recently, whole-genome sequencing has confirmed the enrichment of ID3 mutations across BL subtypes and identified coding and noncoding alterations in several other driver genes, including IGLL5, BACH2, SIN3A, and DNMT1.29 Whole-genome sequencing and transcriptome analyses have also demonstrated that EBV infection occurring in the context of BL results in a specific genomic phenotype, as evidenced by an increased mutational burden and aberrant somatic hypermutation.30 Interestingly, although the 3 variants of BL arise from shared genomic origins, the genomic profiles of sporadic and immunodeficiency-associated BL are more closely related than those seen in endemic BL, presumably because of the uniform presence of EBV infection in the later subtype. These findings raise the question as to whether EBV status provides a more clinically relevant classification than geographic region.29,30
Diagnosis and prognosis
Although there are differences in clinical presentation across BL variants, BL uniformly presents with rapidly progressive disease. Given the benefit of prompt initiation of therapy, biopsies and diagnostic evaluations should be expedited when BL is suspected and enhanced supportive care should be provided. Baseline workup of suspected BL includes imaging, laboratory evaluations, HIV and hepatitis B testing, and a diagnostic lumbar puncture with cytology and flow cytometry of cerebrospinal fluid (CSF) to evaluate for CNS involvement.
Because of the high rates of cell proliferation and turnover, especially in patients with advanced stage or bulky disease, lactate dehydrogenase (LDH) is often elevated at baseline, and spontaneous tumor lysis syndrome (TLS) can occur. TLS, which occurs as the result of release of intracellular contents into the blood, is characterized by hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia.31 If identified, TLS should be considered a medical emergency given the potential for rapid progression of life-threatening complications, including renal failure, cardiac arrhythmias, and seizures. If BL is suspected, prophylactic allopurinol should be promptly initiated, and TLS laboratory values should be monitored closely. TLS can also be induced with treatment initiation, thus requiring aggressive hydration and monitoring. For patients with evidence of spontaneous TLS or those at high risk, defined as stage III/IV disease and/or LDH ≥2 times the upper limit of normal, consensus guidelines recommend the use of rasburicase.32 In cases where patients have high-burden disease, are frail, or are presenting with baseline laboratory abnormalities including renal insufficiency, prephase therapy can be implemented to mitigate risk. Prephase treatment strategies, which typically include the use of prednisone and cyclophosphamide, with or without vincristine, allow for the prevention of fulminant TLS that may be seen with more intensive multiagent therapy.4 In addition, for patients with hyperbilrubinemia related to disease that precludes the administration of anthracyclines or vinca-alkaloids, employing a prephase can be an effective strategy.
Across clinical trials, many patients with BL can be cured with intensive chemotherapy, with 5-year overall survival (OS) rates ranging from 75% to 85% (Table 1). In a recent real-world cohort, the majority of patients were also cured of their disease, although with a slightly more modest 3-year progression-free survival (PFS) and OS of 64% and 70%, respectively.12 Despite these favorable outcomes for many patients, treatment-related mortality (TRM) and risk of relapse remain high in select subgroups of patients. Although there is not a universal prognostic scoring system to delineate risk in BL, a variety of factors have been correlated with outcome. Clear factors that contribute to risk include the age of the patient and their performance status, as older or frailer patients may not be able to tolerate the intensive therapy required for cure. Other variables, including black race, high LDH, advanced stage, and bone marrow or CNS involvement, have also been independently associated with poorer outcome.12,33-35 Access to intensive supportive care is also fundamental to successful therapy. This is demonstrated by the experience in sub-Saharan Africa, where poorer outcomes in the pediatric population are in part due to decreased ability to support patients through high-intensity chemotherapy.4
Treatment options for BL
| Regimen | n | Median age | Elevated LDH, % | TRM, % | EFS/PFS | OS |
|---|---|---|---|---|---|---|
| CODOX-M/IVAC39 | 41 | 25 | 67 | 0 | 2 y EFS 92% | |
| CODOX-M/IVAC40 | 52 | 35 | 63 | 7 | 2 y EFS 65% | 2 y OS 73% |
| CODOX-M/IVAC41 | 53 | 37 | 75 | 8 | 2 y PFS 64% | 2 y OS 67% |
| CALGB43 | ||||||
| Cohort 1 | 52 | 44 | 90 | 11 | 3 y EFS 52% | 3 y OS 54% |
| Cohort 2 | 49 | 50 | 83 | 3 y EFS 45% | 3 y OS 50% | |
| R+CALGB44 | 105 | 44 | 70 | 7 | 2 y EFS 74% | 2 y OS 78% |
| HyperCVAD42 | 26 | 58 | 100 | 19 | 3 y CCR 61% | 3 y OS 49% |
| R+HyperCVAD50 | 31 | 46 | 70 | 0 | 3 y EFS 80% | 3 y OS 89% |
| LMB45 | 72 | 33 | 60 | 4 | 2 y EFS 66% | 2 y OS 70% |
| R+LMB51 | 257 | 47 | 70 | 3 y EFS 75% (R) | 3 y OS 83% (R) | |
| 3 y EFS 65% (no R) | 3 y OS 70% (no R) | |||||
| DA-EPOCH46 | 19 | 25 HIV− | 37 | 0 | EFS 95% | OS 100% |
| SC-EPOCH-RR46 | 11 | 44 HIV+ | 82 | 0 | EFS 90% | OS 100% |
| DA-EPOCH-R47 | 113 | 49 | 69 | 4 | EFS 85% | OS 87% |
| Regimen | n | Median age | Elevated LDH, % | TRM, % | EFS/PFS | OS |
|---|---|---|---|---|---|---|
| CODOX-M/IVAC39 | 41 | 25 | 67 | 0 | 2 y EFS 92% | |
| CODOX-M/IVAC40 | 52 | 35 | 63 | 7 | 2 y EFS 65% | 2 y OS 73% |
| CODOX-M/IVAC41 | 53 | 37 | 75 | 8 | 2 y PFS 64% | 2 y OS 67% |
| CALGB43 | ||||||
| Cohort 1 | 52 | 44 | 90 | 11 | 3 y EFS 52% | 3 y OS 54% |
| Cohort 2 | 49 | 50 | 83 | 3 y EFS 45% | 3 y OS 50% | |
| R+CALGB44 | 105 | 44 | 70 | 7 | 2 y EFS 74% | 2 y OS 78% |
| HyperCVAD42 | 26 | 58 | 100 | 19 | 3 y CCR 61% | 3 y OS 49% |
| R+HyperCVAD50 | 31 | 46 | 70 | 0 | 3 y EFS 80% | 3 y OS 89% |
| LMB45 | 72 | 33 | 60 | 4 | 2 y EFS 66% | 2 y OS 70% |
| R+LMB51 | 257 | 47 | 70 | 3 y EFS 75% (R) | 3 y OS 83% (R) | |
| 3 y EFS 65% (no R) | 3 y OS 70% (no R) | |||||
| DA-EPOCH46 | 19 | 25 HIV− | 37 | 0 | EFS 95% | OS 100% |
| SC-EPOCH-RR46 | 11 | 44 HIV+ | 82 | 0 | EFS 90% | OS 100% |
| DA-EPOCH-R47 | 113 | 49 | 69 | 4 | EFS 85% | OS 87% |
Treatment
With its rapid growth, BL is a highly chemotherapy-sensitive disease and was one of the early cancers in which cures were achieved with chemotherapy alone.36,37 Historically, standard regimens used for DLBCLs, such as CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) with the addition of methotrexate, were associated with high rates of treatment failure.38 Since the late 1980s, intensified multiagent chemotherapy regimens have been successful in the treatment of BL (Table 1).
In 1996, Magrath and colleagues, at the National Cancer Institute (NCI), published their experience with cyclophosphamide, doxorubicin, vincristine, methotrexate, ifosfamide, cytarabine, and etoposide (CODOX-M/IVAC).39 The regimen includes both high-dose, systemically administered and intrathecal methotrexate and cytarabine to treat and prevent disease recurrence in the CNS. Patients with low-risk disease, defined as a single site of disease measuring <10 cm with a normal LDH or completely surgically resected abdominal disease, were treated with 3 cycles of CODOX-M. All other patients (high-risk) received 2 courses of alternating CODOX-M and IVAC (Figure 3). In 21 children and 20 adults with BL with median age of 25, the 2-year event-free survival (EFS) was 92%. Toxicity was predominantly severe myelosuppression resulting in significant rates of infection.
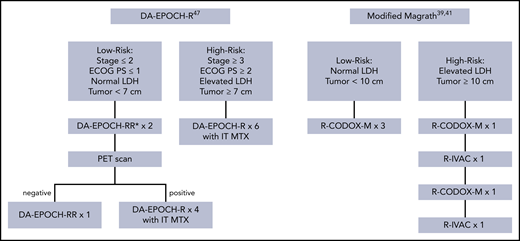
Commonly used treatment approaches for BL. Treatment schemas of commonly used treatment approaches in BL, including DA-EPOCH-R and the modified Magrath regimen. *RR implies patients were treated with rituximab on days 1 and 5. IT MTX, intrathecal methotrexate; PET, positron emission tomography.
Commonly used treatment approaches for BL. Treatment schemas of commonly used treatment approaches in BL, including DA-EPOCH-R and the modified Magrath regimen. *RR implies patients were treated with rituximab on days 1 and 5. IT MTX, intrathecal methotrexate; PET, positron emission tomography.
Results of the Magrath regimen in subsequent studies were less favorable, at least in part, because of increased rates of toxicity in older adults. In a study of 52 patients with median age of 35 treated with CODOX-M/IVAC, OS at 2 years in low- and high-risk patients was 82% and 70%, respectively.40 In this population of patients, only 80% was able to receive full-disease therapy. Adjustments were made to the regimen to reduce toxicity with a decrease in the doses of methotrexate and cytarabine in a subsequent trial.41 PFS), however, was compromised in the high-risk patients at 49% with 85% of low-risk patients achieving long-term disease control.
Intensive pediatric regimens have also been modified for adults with BL. Hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone alternating with high-dose methotrexate and cytarabine (hyper-CVAD), which was developed at the MD Anderson Cancer Center (Houston, TX), was initially adapted from a regimen used to treat acute lymphoblastic leukemia, including Burkitt leukemia.42 The regimen was initially tested in 26 patients with 3-year OS of 49%. TRM was severe at ∼20%, highlighting the challenges of using intensive regimens in older patients. Age was a predictor of outcome with OS of 17% in patients over 60.
The Cancer and Leukemia Group B (CALGB) developed a similar multiagent regimen. Patients received a prephase of cyclophosphamide and prednisone followed by 3 cycles of ifosfamide, methotrexate, vincristine, cytarabine, etoposide, and dexamethasone alternating with cyclophosphamide, methotrexate, vincristine, doxorubicin, and dexamethasone.43 In the first iteration of the trial, patients received 12 doses of intrathecal chemotherapy as well as 2400 cGy of cranial radiation. The trial was amended after 52 patients were treated, given high rates of severe neurotoxicity. Subsequently, itrathecal doses were reduced to 7 and radiotherapy was administered only to patients with baseline bone marrow involvement. Overall, 92 patients were treated with 5-year OS of 52%. In a subsequent study with the addition of rituximab, outcomes improved with EFS and OS at 2 years of 74% and 78%, respectively.44 Treatment-associated mortality occurred in 10 patients in the first study and 7 patients in the second.
In Europe, adapted pediatric acute lymphoblastic leukemia regimens have also been employed for the treatment of adults with BL. In the French lymphome malin B (LMB) regimen, patients were assigned to low-risk (group A: resected stage I and abdominal stage II disease), intermediate-risk (group B: neither low- nor high-risk), and high-risk (group C: bone marrow and/or CNS involvement) groups.45 Group A was treated with cyclophosphamide, doxorubicin, vincristine, and prednisone (COPAD). Groups B and C received prephase therapy with cyclophosphamide, vincristine, and low-dose steroids to debulk disease and reduce the risk of tumor lysis. Additional treatment of group B consisted of 5 cycles of therapy, including high-dose methotrexate, cytarabine (COPADM/CYM), and intrathecal methotrexate. Group C received 8 cycles with increased doses of methotrexate, cytarabine, etoposide (COPADM, CYVE), and intrathecal therapy with methotrexate and cytarabine. Patients with CNS involvement received cranial radiotherapy to 24 Gy. In 72 patients with median age of 33 treated with this approach, the 2-year EFS and OS were 65% and 70%, respectively. Three patients died of treatment-related toxicity. Age over 32 and elevated LDH were associated with inferior survival.
The infusion chemotherapy regimen dose-adjusted rituximab, etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin (R-EPOCH) was designed by investigators at the 1nts, including 11 with HIV, were treated in an initial trial with median age of 33 for the entire cohort and 25 for the HIV−patients.46 LDH was elevated in 53% of patients, and 10% had high-risk disease, defined as bone marrow and/or CNS involvement. Patients were treated with 2 cycles beyond complete remission (6 to 8 total) with the exception of HIV patients who received 1 cycle beyond complete response (3 to 6 total) with 2 doses of rituximab administered with each cycle. CNS-directed therapy consisted of 8 doses of prophylactic intrathecal methotrexate with additional doses for patients with leptomeningeal disease. With long-term follow-up of >6 years, freedom from progression and OS were 95% and 100%, respectively. No patients with HIV experienced progression, and OS was 90%. Rates of febrile neutropenia were low, and there were no cases of TRM.
A confirmatory study in the multicenter setting was recently published, in part to address criticism that the study conducted at the NCI included a relatively favorable group of patients.47 Low-risk patients received 3 cycles of dose-adjusted R-EPOCH without CNS prophylaxis, and high-risk patients received 6 cycles with intrathecal CNS prophylaxis or extended intrathecal treatment if the leptomeninges were involved (Figure 3). One hundred thirteen patients, 28 of whom were HIV+, with median age of 49 were treated. Thirteen percent were defined as low risk, and 28 had baseline bone marrow or peripheral blood involvement. Eleven had CSF disease.47 With median follow-up of ∼5 years, the EFS and OS were 84.5% and 87%, respectively, with 100% EFS in low-risk patients. Regarding patients with leptomeningeal disease, however, EFS was 45.5%, and EFS for patients with either bone marrow or CSF involvement was 58.6% compared with 92.4% with neither. TRM was 4%.
There are no published randomized trials in BL comparing chemotherapy regimens. A recent retrospective comparison of patients treated in Europe demonstrated no difference in outcomes in 105 patients treated with one of 4 regimens: LMB, BFM, HOVON, and CODOX-M/IVAC with 5-year PFS of 69%.48 On cost analysis and treatment duration, CODOX-M/IVAC was associated with the most favorable profile. An ongoing European trial is comparing R-CODOX-M/IVAC to dose-adjusted R-EPOCH. In the United States, a recent retrospective analysis demonstrated that CODOX-M/IVAC, hyper-CVAD, and R-EPOCH were the most commonly used regimens across 30 centers without clear differences in outcome.12 Based on the existing data, for patients without bone marrow or CNS involvement, outcomes with R-EPOCH appear very favorable with possibly less toxicity compared with regimens incorporating high-dose methotrexate and cytarabine. The appropriate treatment likely requires evaluation of a variety of factors, including the age and performance status of the patient, as well as the presence of high-risk features, such as CNS involvement.
Rituximab
Given the improvement in outcomes with its use in aggressive lymphoma, rituximab has been incorporated broadly into BL regimens. Compared with historical controls, the inclusion of rituximab is associated with improvements in outcomes with the CODOX-M/IVAC and hyper-CVAD regimens.49,50 In a randomized study employing the LMB chemotherapy backbone in 260 HIV− patients with intermediate (no CNS or marrow involvement) or high-risk (with either) disease, patients were randomized to receive rituximab plus their standard risk-stratified regimen vs chemotherapy alone.51 With a median follow-up of 38 months, EFS and OS were superior in the rituximab-containing arms at 75% vs 62% and 83% vs 70%, respectively. Adverse events were similar between the 2 arms.
CNS involvement
CNS involvement has clearly been identified as a poor prognostic factor in BL across multiple studies.12,47 The risk of CNS involvement in BL is high, with up to 20% of patients presenting with CNS involvement at the time of diagnosis, with leptomeningeal involvement being more common than parenchymal disease.12 In the absence of prophylaxis, 30% to 50% will develop CNS disease.52 Given these findings, incorporation of CNS-directed prophylaxis and therapy is a fundamental component of BL therapy, although the optimal strategy to perform this remains unclear. Most BL regimens employ intrathecal chemotherapy in combination with high-dose methotrexate and/or cytarabine, which could cross the blood-brain barrier. Other regimens, such as R-EPOCH, incorporate intrathecal chemotherapy alone.
Although options for CNS prophylaxis and therapy have not been directly compared, systemic therapy is often preferred for patients with CNS involvement. In the recent multicenter trial of R-EPOCH, many patients with CNS involvement had poor outcomes, although the sample size was small in this study. This study did, however, demonstrate that intrathecal prophylaxis can successfully prevent CNS relapse in high-risk patients without baseline involvement, with a relapse rate of only 2% in this population.47 Therefore, for patients with CNS involvement, CODOX-M/IVAC may be a preferred option in a patient eligible for intensive therapy.
BL in older patients
The effectiveness of intensive therapies for BL must be balanced by the potential for TRM, particularly in older patients or those with underlying comorbidities. In a large retrospective analysis of patients with BL, TRM was 13% to 17% in patients >60 years, regardless of treatment regimen.12 The most common cause of death was sepsis. TRM was higher in patients receiving hyper-CVAD compared with CODOX-M/IVAC or DA-EPOCH-R. In the recent prospective study of R-EPOCH, the median age was 49 years, with >62% of patients who were ≥40 years of age, highlighting the feasibility of this regimen for older patients.47
Role of stem cell transplantation
Consolidation with stem cell transplantation using BEAM (carmustine, etoposide, cytarabine, and melphalan) conditioning was examined in a small study of 27 patients after 2 cycles of cyclophosphamide, doxorubicin, etoposide, mitoxantrone, and prednisone.53 A large retrospective analysis of use of transplant from 1985 to 2007 from the Center for International Blood and Marrow Transplant Research demonstrated a decline in the use of autologous transplant (n = 113) over time.54 Five-year OS was 83% for patients undergoing transplant in first remission compared with 31% in all other patients. One hundred twenty-eight patients underwent allogeneic transplant with 10% of patients after 1 line of therapy with 5-year PFS of 27%.
Relapsed and refractory disease
The prognosis for patients with relapsed or refractory BL is poor. In a single-center experience of 145 patients with BL or high-grade B-cell lymphoma treated with hyper-CVAD, 35 patients with relapsed or refractory disease were reported.55 Thirty-nine percent of patients responded to second-line therapy. The median OS was 2.8 months, and only 2 patients were alive at 48 months. In 157 children and adolescent patients with relapsed/refractory BL treated with BFM-type initial therapy between 1986 and 2016, 3-year OS was 18.5%. Rituximab plus infusional second therapy followed by allogeneic transplant was associated with 67% survival compared with 18% with all other approaches.56
Novel approaches and future directions
Novel therapeutic strategies are urgently needed for BL, particularly in patients who are unable to tolerate intensive therapies or those with relapsed disease. Clinical trials have been challenging in adults with BL, because of both the rarity and the aggressive nature of the disease, often with urgent need for treatment initiation. CD19-directed chimeric antigen receptor T-cell therapy has not been systematically assessed in patients with BL, although a single case report describes complete metabolic response in a heavily pretreated patient who had undergone prior allogeneic stem cell transplantation.57 Unfortunately, the patient subsequently died of treatment-related complications after undergoing haploidentical transplant. Similarly, bispecific antibodies that simultaneously target B cells and T cells have shown great promise in B-cell NHLs, although require further evaluation in BL. Genomic alterations in BL, including those that impact BCR/phosphatidylinositol 3-kinase signaling as well as cell-cycle regulation, provide rationale for preclinical studies of targeted therapeutic approaches as well.
Conclusions
BL is a highly chemotherapy-sensitive disease. Intensified combination chemotherapy approaches with rituximab yield OS rates of ∼75% to 85%. Age is an important predictor of outcome, as TRM is high in older patients. Dose-adjusted R-EPOCH is a generally more tolerable combination, although in patients with baseline CNS disease, the lack of high-dose systemically administered therapy likely contributes to worse outcomes. Therapy for patients with primary refractory or relapsed disease represents an unmet medical need. Studies using chimeric antigen receptor T-cell cells, bispecific antibodies, and other novel approaches are underway.
Authorship
Contribution: J.C. and A.L. contributed equally to the writing of the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ann LaCasce, Dana-Farber Cancer Institute, 450 Brookline Ave, M227, Boston, MA 02115; e-mail: ann_lacasce@dfci.harvard.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal