

In this issue of Blood, present a custom workflow using a 110 gene panel with the Mission Bio Tapestri platform to investigate clonal evolution and heterogeneity in human T-cell acute lymphoblastic leukemia (T-ALL).1
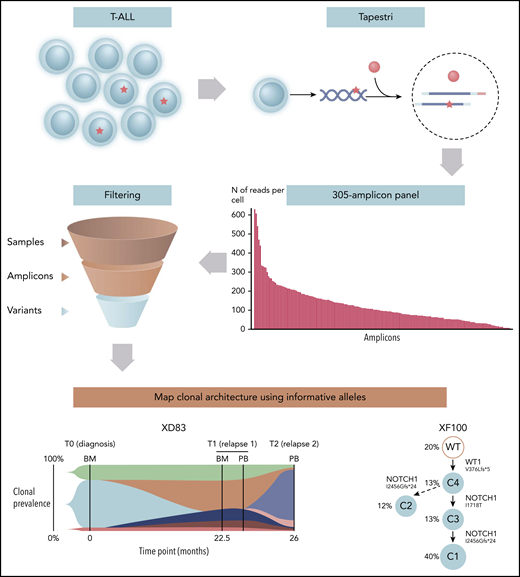
Single-cell genomic analysis of T-ALL. Genomic DNA from single T-ALL cells are isolated and barcoded using the Mission Bio Tapestri platform. Alleles are sequenced by NGS using a bespoke 305-amplicon panel for 110 genes on PCR-amplified barcoded genomic DNA. Informative alleles are identified after sample and amplicon filtering, identification of allele dropouts (ADOs), and variant curation. T-ALL single-cell clonal architecture is reconstructed. BM, bone marrow; PB, peripheral blood; WT, wild type.
Single-cell genomic analysis of T-ALL. Genomic DNA from single T-ALL cells are isolated and barcoded using the Mission Bio Tapestri platform. Alleles are sequenced by NGS using a bespoke 305-amplicon panel for 110 genes on PCR-amplified barcoded genomic DNA. Informative alleles are identified after sample and amplicon filtering, identification of allele dropouts (ADOs), and variant curation. T-ALL single-cell clonal architecture is reconstructed. BM, bone marrow; PB, peripheral blood; WT, wild type.
Single-cell analysis has provided significant insights into genomic variation of cancer at the clonal level. This approach was originally employed to detect copy-number variation and single-nucleotide variants in a colorectal cancer cell line2 and breast cancer3 and allowed reconstruction of genomic events based on the clonal distribution of genomic alterations.
A number of methods for characterizing clonal heterogeneity have been used in the genomic analysis of ALL. These include bulk and single-cell analyses that have incorporated fluorescence in situ hybridization (FISH) with microarray, conventional, and next-generation sequencing (NGS). Initial approaches combined bulk sequencing with targeted microfluidic resequencing for variant confirmation and reconstruction of clonal architecture for analysis of clonal heterogeneity in ETV6-RUNX1 leukemia.4 A single-nucleotide polymorphism array for copy-number alterations and 4-gene multiplex targeted quantitative polymerase chain reaction (qPCR) were implemented for investigation of B-cell ALL,5 whereas clonal analysis of STIL-TAL1+ T-cell ALL6 used a combination of multiplex targeted qPCR and FISH.
Workflows for NGS single-cell genomic analysis depend on PCR-based whole-genome amplification of picogram amounts of DNA isolated from single cells using fluidic platforms. The workflow for the Mission Bio Tapestri platform for single-cell DNA extraction followed by a targeted amplicon panel for NGS sequencing has been recently described for characterization of the genetic heterogeneity of acute myeloid leukemia7 and investigation of the clonal basis for therapy resistance to gilteritinib.8
Albertí-Servera et al present a single-cell DNA amplicon NGS sequencing method using PCR amplification and a custom 110-gene panel (305 amplicons; see figure) and workflow using the Tapestri platform for cell encapsulation and barcoding to investigate clonal evolution and heterogeneity in human T-ALL. Their report advances the approach through the application and validation of a custom amplicon panel and bioinformatic workflow incorporating the Mission Bio Pipeline with a process for identifying variant allele frequencies from single-cell sequencing data. Importantly, this includes a method that was validated by the group to estimate and identify ADO. This technical limitation of single-cell genomic analysis is of particular importance, because at limiting concentrations of DNA, insufficient allele amplification may result in the PCR product/allele ratio not accurately reflecting that of the original genomic DNA. Hence, loss of heterozygosity may be inaccurately identified through ADO (ie, a false positive).
The data presented by Albertí-Servera et al in the analysis of 25 samples from 8 pediatric T-ALL patients demonstrate that high-quality data can be produced. Of note, although sequencing reads were uneven across the 305 amplicons, a vast majority were informative, with an acceptable median ADO rate of ∼9%. After variant filtering, an interquartile range of 20 to 29 good-quality variants were identified per patient.
From these data, clonal analysis was undertaken to compare blood vs bone marrow sampling and the clonal composition and architecture of all leukemias in which informative alleles were identified. Their study identified heterogeneous NOTCH1 mutations as secondary subclonal events, as this group had previously noted at a single-cell level,9 as well as cooccurring mutations in signaling pathways. Patterns of clonal evolution during treatment and relapse were also able to be described.
As we learn more about the clonal heterogeneity of cancer, it will be increasingly important to validate single-cell approaches for genomic analysis. Optimization of PCR conditions, design and validation of amplicon panels and bioinformatic workflows, including the assessment of thresholds that are applied for amplicon filtering and variant allele and heterozygosity curation, will all be needed. Needless to say, ongoing accumulation of data to identify emergent patterns in disease behavior and treatment resistance will also be required before the promise of these technologies can be realized and eventually help to refine our clinical approach; assisting in the application of conventional and novel targeted therapies to an individual patient.
Although the insights gained by this report by Albertí-Servera et al into disease pathogenesis and therapy response were perhaps not revolutionary, the comprehensive and validated methods undertaken and described provide a robust roadmap for single-cell genomic analysis, including identification of the potential pitfalls in this approach.
Conflict-of-interest disclosure: The author declares no competing financial interests.
