


Abstract
Mutations of the nucleophosmin (NPM1) gene, encoding for a nucleolar multifunctional protein, occur in approximately one-third of adult acute myeloid leukemia (AML). NPM1-mutated AML exhibits unique molecular, pathological, and clinical features, which led to its recognition as distinct entity in the 2017 World Health Organization (WHO) classification of myeloid neoplasms. Although WHO criteria for the diagnosis of NPM1-mutated AML are well established, its distinction from other AML entities may be difficult. Moreover, the percentage of blasts required to diagnose NPM1-mutated AML remains controversial. According to the European LeukemiaNet (ELN), determining the mutational status of NPM1 (together with FLT3) is mandatory for accurate relapse-risk assessment. NPM1 mutations are ideal targets for measurable residual disease (MRD) monitoring, since they are AML specific, frequent, very stable at relapse, and do not drive clonal hematopoiesis of undetermined significance. MRD monitoring by quantitative polymerase chain reaction of NPM1-mutant transcripts, possibly combined with ELN genetic-based risk stratification, can guide therapeutic decisions after remission. Furthermore, immunohistochemistry can be very useful in selected situations, such as diagnosis of NPM1-mutated myeloid sarcoma. Herein, we present 4 illustrative cases of NPM1-mutated AML that address important issues surrounding the biology, diagnosis, and therapy of this common form of leukemia.
Introduction
Acute myeloid leukemia (AML) carrying mutations of nucleophosmin (NPM1), a gene encoding for a multifunctional nucleolar protein with chaperone and shuttling features,1 accounts for ∼30% of adult AML and exhibits distinctive molecular and clinicopathological features.2,3 The aberrant cytoplasmic dislocation of mutant NPM1 is thought to play a key role in leukemogenesis.4,5 NPM1 mutations are infrequent (6.5%) in children,6 peak at middle age, and tend to decrease in patients >70 years, although they can still be detected in ∼20% of older AML patients.7
Following its discovery in 2005,2 it has been a long road before NPM1-mutated AML was recognized by the World Health Organization (WHO) classification of hematopoietic neoplasms as provisional entity in 2008 and then as distinct entity in 2017.8 According to the current WHO classification, NPM1-mutated AML (together with CEBPA-mutated AML) is 1 of 2 AML entities defined by a single gene mutation, although it is likely that this list will grow to include other genes. Because NPM1-mutated AML is relatively frequent, the WHO category of AML with recurrent genetic abnormalities has now approximately doubled in size, with ∼60% to 65% of AML being recognizable by genetic assays. Moreover, simple and low-cost techniques, including immunohistochemistry (IHC) and basic molecular assays, are available for the detection of NPM1 mutations, facilitating the diagnosis of NPM1-mutated AML worldwide,9 which is a major scope of the WHO classification. We also believe that the recognition of NPM1-mutated AML as distinct entity encourages the design of genetic-based clinical trials. The main features of NPM1-mutated AML are summarized in Table 1.
Main characteristics of NPM1-mutated AML
| Specific characteristics and comments |
|---|
| Approximately 30-35% of adult AML (50-60% of AML with normal cytogenetic). Less frequent in children (∼2-8%).* Female predominance. |
| BM usually markedly hypercellular. Mostly myelomonocytic (FAB M4) and monocytic (FAB M5), but all FAB categories are represented. |
| Approximately 23% of cases show multilineage dysplasia. |
| WBC count may be influenced by FLT3 mutational status, progressively increasing from FLT3 wild-type to FLT3-ITDhigh. |
| Frequent association with extramedullary involvement, especially skin (easily detectable by IHC). |
| No/low expression of CD34. The rare CD34+ leukemic cells carry the NPM1 mutation. CD34 positivity has been associated with adverse prognosis. |
| Excellent response to induction chemotherapy. |
| Relatively good outcome in the absence of FLT3-ITD. Prognosis may vary depending upon concomitant mutations. |
| Specific characteristics and comments |
|---|
| Approximately 30-35% of adult AML (50-60% of AML with normal cytogenetic). Less frequent in children (∼2-8%).* Female predominance. |
| BM usually markedly hypercellular. Mostly myelomonocytic (FAB M4) and monocytic (FAB M5), but all FAB categories are represented. |
| Approximately 23% of cases show multilineage dysplasia. |
| WBC count may be influenced by FLT3 mutational status, progressively increasing from FLT3 wild-type to FLT3-ITDhigh. |
| Frequent association with extramedullary involvement, especially skin (easily detectable by IHC). |
| No/low expression of CD34. The rare CD34+ leukemic cells carry the NPM1 mutation. CD34 positivity has been associated with adverse prognosis. |
| Excellent response to induction chemotherapy. |
| Relatively good outcome in the absence of FLT3-ITD. Prognosis may vary depending upon concomitant mutations. |
A synonym of NPM1-mutated AML is “AML with cytoplasmic nucleophosmin” (NPM1c+).
FAB, French-American-British.
Likely because NPM1 mutations are often preceded by clonal hematopoiesis, which is very rare in children.
According to the 2017 WHO classification,8 diagnosis of NPM1-mutated AML requires ≥20% of blasts. Another critical diagnostic step is the identification of the exact type of NPM1 mutation by sequencing.11 This information is required to set up real-time quantitative polymerase chain reaction (RT-qPCR) for measurable residual disease (MRD) monitoring.10 Detection of cytoplasmic NPM1 by IHC12 may serve as surrogate for molecular diagnosis in cases of dry tap, bone marrow (BM) necrosis,13 or myeloid sarcoma. Moreover, IHC enables detection of all clinically relevant NPM1 mutations (even rare mutations occurring outside exon 12) and is feasible in developing countries not equipped for molecular studies.9
Criteria for distinguishing NPM1-mutated AML from other distinct AML entities are well defined.8 NPM1-mutated AML must also be separated from 2 provisional entities of the 2017 WHO classification: AML with BCR-ABL1 and AML with RUNX1 mutations. When NPM1 mutation and BCR-ABL1 cooccur, the case should be diagnosed as NPM1-mutated AML. However, the addition of an ABL kinase inhibitor to the treatment should be considered. Similarly, when NPM1 and RUNX1 mutations coexist (<1% cases), the patient should classified as NPM1-mutated AML.8 Accordingly, RUNX1 mutations occurring in NPM1-mutated AML are quite different from those classically found in other AMLs, since they are in frame, located outside the RUNT domain, germline, and of unclear functional significance.14
At the time we discovered NPM1 mutations in AML, we also reported for the first time that NPM1 and FLT3 internal tandem duplication (ITD) mutations were frequently associated, suggesting that they were mechanistically related.1 Several investigators have subsequently contributed to demonstrate the biological and clinical importance of this association to the extent that the status of NPM1 and FLT3 genes is now among the most important pillars upon which the European LeukemiaNet (ELN) prognostication system is built.15 The 2017 ELN guidelines15 (as compared with the 2008 version) recognize 3 (instead of 4) genetic-based risk groups: favorable, intermediate, and high. Another substantial revision has been the introduction of FLT3-ITD allelic ratio determined as the ratio of the area under the curve of FLT3-ITD and FLT3 wild-type. NPM1-mutated AML without FLT3-ITD or with FLT3-ITDlow (ratio < 0.5) are classified as favorable-risk categories while NPM1-mutated AML with FLT3high (ratio > 0.5) is regarded as intermediate risk.15
Selection of the best treatment strategy for patients with NPM1-mutated AML can be potentially refined combining the ELN prognostication model with the results of MRD evaluated by quantifying NPM1mut transcript copies10 at diagnosis and at selected time points after therapy. In fact, NPM1 mutations are ideal targets for MRD monitoring, since they are AML specific, frequent, and stable at relapse, and, unlike DNMT3A, TET2, or IDH mutations, they do not drive clonal hematopoiesis, being a “gatekeeper” for AML.16-18
Although therapies specifically directed against the NPM1 mutant or downstream signaling pathways are not yet available, recent clinical studies have identified drug combinations that may be particularly effective in NPM1-mutated AML (eg, venetoclax-based regimens).19
Herein, we present 4 illustrative cases of NPM1-mutated AML, with the aim to address relevant issues on the diagnosis and therapy of this frequent leukemia.
Scenario 1: Young adult patient with NPM1-mutated AML, FLT3 wild-type, and skin involvement
A 60-year-old man presented with multiple skin nodules without symptoms. A complete blood count (CBC) showed white blood cell (WBC) 1.6 × 109/L, hemoglobin (Hb) 13.8 g/dL, and platelets 167 × 109/L. BM was diffusely infiltrated by myeloid blasts expressing cytoplasmic NPM1 at IHC. NPM1 mutation A and FLT3 wild-type were detected; karyotyping was normal. Skin biopsy specimen showed leukemic involvement (Figure 1A-C). A “7+3” induction chemotherapy (CHT) led to complete remission (CR) and disappearance of all skin lesions (Figure 1D-E). RT-qPCR showed 3.9-log reduction of NPM1mut transcripts in BM following induction, 4.5-log reduction after the first consolidation, and MRD negativity after the second consolidation cycle. In the following 6 months, the NPM1mut transcripts in BM progressively increased (Figure 1F). He received pre-emptive treatment with off-label dactinomycin,20 achieving a slight MRD decrease (Figure 1F), followed by allogeneic hematopoietic stem cell transplantation (allo-HSCT)21 from an haploidentical donor. He is now in molecular CR (Figure 1F) >3 years after the allotransplant.
![NPM1-mutated myeloid sarcoma of the skin. (A) Dermal infiltration by leukemic cells (skin biopsy, B5-fixed; hematoxylin-eosin, original magnification ×400). (B) Leukemic cells exhibit aberrant cytoplasmic expression of NPM1 (arrow). Normal cells of epidermis show nucleus-restricted expression of NPM1 (double arrows) (immune alkaline phosphatase anti-alkaline phosphatase [APAAP] staining, original magnification ×400). (C) Leukemic cells in the derma are myeloperoxidase-positive (APAAP staining, original magnification ×400). Ep, epidermis. (D-E) Cutaneous nodules at diagnosis (D) disappeared after induction chemotherapy (E). (F) Monitoring of NPM1mut transcripts during therapy and follow-up (see text). 0.0001 is equivalent to MRD negativity. ActD, dactinomycin; Rel, relapse; RQ-PCR, RT-qPCR.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/5/10.1182_blood.2020008211/1/m_bloodbld2020008211f1.png?Expires=1769102067&Signature=lcabVCF53RkEeTMyisgK3ij3Ta0K6r3w26JFYaYsWh6EML1jAJszDMnW5xBoDi9xXCU1W~AykjeRzWOmO-O5pmGOnfUhwYNZz8l6Uc07p5QIKDJO-M1tGUMq9KhpUyB4IH1h26S~m6MWWGBwO7y6ywkb3dN3a6CrBeW5ZSrNEHL5MAnQWtwP4jnYHF9nSvQ~yF5fQL2qUdMyBz03UT1~oCoE4x2Pnr9-fKOSmMlVbqk681Z4nHqy9N0PSf8v-2aZbqOF9pQEUuCAWzy4E6MldTc5UdERzbEp8QNe0W~6pFf2gbL0xsr8CkdXAdAtTuNHLouNLqVb~XoMDlWH-JLlMA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
NPM1-mutated myeloid sarcoma of the skin. (A) Dermal infiltration by leukemic cells (skin biopsy, B5-fixed; hematoxylin-eosin, original magnification ×400). (B) Leukemic cells exhibit aberrant cytoplasmic expression of NPM1 (arrow). Normal cells of epidermis show nucleus-restricted expression of NPM1 (double arrows) (immune alkaline phosphatase anti-alkaline phosphatase [APAAP] staining, original magnification ×400). (C) Leukemic cells in the derma are myeloperoxidase-positive (APAAP staining, original magnification ×400). Ep, epidermis. (D-E) Cutaneous nodules at diagnosis (D) disappeared after induction chemotherapy (E). (F) Monitoring of NPM1mut transcripts during therapy and follow-up (see text). 0.0001 is equivalent to MRD negativity. ActD, dactinomycin; Rel, relapse; RQ-PCR, RT-qPCR.
NPM1-mutated myeloid sarcoma of the skin. (A) Dermal infiltration by leukemic cells (skin biopsy, B5-fixed; hematoxylin-eosin, original magnification ×400). (B) Leukemic cells exhibit aberrant cytoplasmic expression of NPM1 (arrow). Normal cells of epidermis show nucleus-restricted expression of NPM1 (double arrows) (immune alkaline phosphatase anti-alkaline phosphatase [APAAP] staining, original magnification ×400). (C) Leukemic cells in the derma are myeloperoxidase-positive (APAAP staining, original magnification ×400). Ep, epidermis. (D-E) Cutaneous nodules at diagnosis (D) disappeared after induction chemotherapy (E). (F) Monitoring of NPM1mut transcripts during therapy and follow-up (see text). 0.0001 is equivalent to MRD negativity. ActD, dactinomycin; Rel, relapse; RQ-PCR, RT-qPCR.
Questions and recommendations
The first question is what the optimal frontline therapy for our patient should be. Young adults (≤60 years) with NPM1-mutated AML without FLT3-ITD should receive induction plus consolidation therapy15,22-25 (Figure 2). Based on recent studies, the addition of gemtuzumab ozogamicin (GO) to CHT may be of benefit in NPM1-mutated AML. A meta-analysis26 clearly showed a survival benefit for patients with intermediate-risk cytogenetics and NPM1-mutated AML due to a reduced relapse risk. This was confirmed in the ALFA0701 trial27 that also proved the role of GO in serially reducing NPM1mut transcripts. The AMLSG study also showed an improved relapse-free survival28 and a reduced cumulative incidence of relapse in NPM1-mutated AML due to deeper reduction of NPM1mut transcript levels across all treatment cycles.29 However, the early primary end point of this study (event-free survival) was not met due to excessive toxicity, possibly related to a combination of the dosing strategy and the inclusion in the regimen of ATRA and etoposide.28 GO benefit in this study was mostly observed in females ≤70 years without FLT3-ITD.28 Altogether, these findings support the incorporation of GO into the frontline treatment of NPM1-mutated AML.

Treatment algorithm for young fit NPM1-mutated AML patients with FLT3-ITDwt, FLT3-ITDlow, and FLT3-ITDhigh. All patients with mutated FLT3 (irrespective of allelic ratio) should receive FLT3 inhibitor plus CHT as induction/consolidation therapy. Some authorities consider allo-HSCT in CR1 for patients with FLT3-ITDlow, except for cases with ratio <0.1. *Whether patients with suboptimal reduction of NPM1 transcripts after 2 CHT cycles with or without FLT3 inhibitor (depending on FLT3 status) may benefit from allo-HSCT should be investigated in prospective clinical trials. **Proceed to allo-HSCT as soon as possible. ^Whether the subset of patients with FLT3-ITDhigh achieving MRD negativity after 2 CHT cycles may avoid allo-HSCT in CR1 should be investigated in prospective clinical trials. FLT3i, FLT3 inhibitor; NRM, nonrelapse mortality; WT, wild-type.
Treatment algorithm for young fit NPM1-mutated AML patients with FLT3-ITDwt, FLT3-ITDlow, and FLT3-ITDhigh. All patients with mutated FLT3 (irrespective of allelic ratio) should receive FLT3 inhibitor plus CHT as induction/consolidation therapy. Some authorities consider allo-HSCT in CR1 for patients with FLT3-ITDlow, except for cases with ratio <0.1. *Whether patients with suboptimal reduction of NPM1 transcripts after 2 CHT cycles with or without FLT3 inhibitor (depending on FLT3 status) may benefit from allo-HSCT should be investigated in prospective clinical trials. **Proceed to allo-HSCT as soon as possible. ^Whether the subset of patients with FLT3-ITDhigh achieving MRD negativity after 2 CHT cycles may avoid allo-HSCT in CR1 should be investigated in prospective clinical trials. FLT3i, FLT3 inhibitor; NRM, nonrelapse mortality; WT, wild-type.
The second question refers to the best postremission therapy in cases like our patient. Allo-HSCT in first CR (CR1) is not recommended for NPM1-mutated AML patients without FLT3-ITD.30 Allo-HSCT has been advocated for patients ≤50 years with low predicted transplant-related mortality and an HLA-identical donor.31 However, allotransplant results in better relapse-free survival, but not overall survival (OS), likely because patients can be salvaged by allo-HSCT in second CR.31 A suboptimal reduction of NPM1 transcripts after 2 CHT cycles could potentially help selecting patients with favorable genotype who may benefit from allo-HSCT, but this needs to be prospectively validated. Under these circumstances, we usually monitor the MRD, and in case of progressive increase of NPM1mut transcripts over time, we consider administering CHT in the attempt to reduce MRD before proceeding to allo-HSCT.
A yet-controversial issue remains whether patients with NPM1-mutated AML with FLT3-ITDlow, which also belongs to the ELN favorable-risk category, should undergo allo-HSCT in CR1, as a good outcome of this genotype has been recently questioned in several studies. Straube et al32 found a lower survival and increased relapse risk compared with other favorable-risk patients. Similarly, Sakaguchi et al33 reported that NPM1-mutated AML patients with FLT3-ITDlow treated with CHT alone did not show favorable outcome (5-year OS, 41.3%) unless they underwent allo-HSCT in CR1. Conversely, Dohner et al,25 in a retrospective analysis of the impact of NPM1/FLT3-ITD genotypes in the RATIFY trial (CHT vs CHT + midostaurin), confirmed the favorable prognosis of NPM1-mutated AML with FLT3-ITDlow treated with CHT + midostaurin (5-year OS, 73%)25 and found no benefit of allo-HSCT in this setting. Indeed, using the Simon-Makuch plots, the authors found that allo-HSCT was beneficial only in the ELN adverse risk group. Overall, these findings suggest that the impact of FLT3-ITDlow in risk stratification may depend on the treatment setting (eg, FLT3 inhibitors). However, some authorities still consider allo-HSCT in CR1, except for patients with a very low FLT3-ITD allelic ratio (eg, <0.1). In FLT3-ITDlow patients treated with standard CHT plus a FLT3 inhibitor, we follow the same policy as in FLT3 wild-type patients. The decisional algorithm for NPM1-mutated AML without FLT3-ITD and with FLT3-ITDlow is shown in Figure 2.
Based on patient’s age and the lack of an HLA-identical donor, we decided not to allotransplant our patient in CR1. Another factor that guided our clinical decision to avoid transplant was the MRD negativity achieved at the end of treatment. Indeed, achieving MRD negativity by RT-qPCR in peripheral blood (PB)34 or BM35 or deep reduction in NPM1mut transcripts36-38 (optimal thresholds remain uncertain39 ) at various time points after CHT predicts favorable outcomes. MRD evaluation in PB after 2 CHT cycles appears even more precise than genetic-based prognostication in predicting risk of relapse.34 Thus, as recommended by ELN,40 we monitored the level of NPM1mut transcripts both in BM and PB every 3 months. This time schedule is based on the observation that kinetics of NPM1-mutated AML is quicker than that of CBF-MYH11 in core-binding factor AML.41 Progressive MRD increase predicts hematological relapse.35,40,42 Unfortunately, despite the favorable genotype and the excellent molecular response, our patient showed a progressive increase in NPM1mut transcripts yet remained in hematological CR. T lymphocytes reactive against tumor-specific HLA-presented peptides of NPM1 mutants have been demonstrated in NPM1-mutated AML patients.43 Relapse in patients achieving MRD negativity may reflect the inability of the immune system to clear residual leukemic cells (below the threshold of detection by RT-qPCR).
NPM1-mutated AML patients in molecular relapse should be offered an allo-HSCT. Some clinicians wait for hematological relapse while others either proceed directly to allo-HSCT or administer pre-emptive therapy44,45 with the aim of reducing MRD levels before allo-HSCT, since MRD levels correlate with allotransplant outcomes.46-48 For this reason, we pre-emptively treated our patient with dactinomycin20 before allo-HSCT. Other options for pre-emptive therapy in NPM1-mutated AML include 5-azacitidine,49 venetoclax,50 and immunotherapy.51 In one study, patients in molecular relapse that received pre-emptive 5-azacitidine showed lower incidence or delayed hematological relapse compared with historical controls.49 In another study, NPM1-mutated AML patients without FLT3-ITD or with FLT3-ITDlow did better if treated at molecular failure rather than at hematological relapse (80% vs 40%, 2-year OS, respectively).45 However, more studies are required to establish the best therapeutic strategy in the setting of molecular relapse.
Another important point highlighted in this case refers to the diagnosis and clinical significance of skin involvement in NPM1-mutated AML. Immunohistochemical detection of cytoplasmic NPM1 helps establishing leukemic skin involvement, a relatively common event in NPM1-mutated AML.52 Based on a large study showing no prognostic significance of extramedullary disease in AML (although mutations were not analyzed),53 we do not regard skin involvement at presentation as a poor prognostic factor in NPM1-mutated AML. Thus, clinical decisions in our patient were exclusively based on ELN risk stratification and MRD response.
Scenario 2: Young adult patient with NPM1-mutated AML and high FLT3-ITD allelic ratio (FLT3-ITDhigh)
A 58-year-old man presented with fatigue, gingival hyperplasia. and bleeding. CBC showed WBC 52.6 × 109/L, Hb 9.4 g/dL, and platelets 48 × 109/L. BM examination was consistent with NPM1-mutated AML, as proven by expression of cytoplasmic NPM1. Diagnosis was confirmed by the identification of NPM1 mutation A and FLT3-ITD (ratio 0.9). The BM karyotype was normal. The patient received a 7+3 induction regimen, achieving CR and only a 1.7-log reduction of NPM1mut transcripts, with FLT3-ITD still detectable with a ratio of 0.01. An additional 2.5-log reduction of NPM1mut copies and FLT3-ITD negativity was achieved after 2 consolidation cycles. The patient underwent a haploidentical HSCT. He is now in molecular CR, 5 years after the allotransplant.
Questions and recommendations
Currently, NPM1-mutated AML patients with FLT3-ITDhigh (ratio ≥ 0.5) should receive conventional chemotherapy plus a FLT3 inhibitor25,54 (not approved at the time our patient was treated) (Figure 2). As discussed in scenario 1, these patients may also benefit from GO incorporation into frontline therapy.27
Allo-HSCT in CR1 represents the best therapeutic option in NPM1-mutated AML patients with FLT3-ITDhigh, like our case15,31,38,55-58 (Figure 2), although some authors claim it may be questionable in patients achieving postremission NPM1mut MRD negativity.59 Our policy is to go to allo-HSCT as soon as possible. If >3-log reduction of NPM1mut in BM is achieved after 1 or 2 CHT cycles, we usually proceed to myeloablative allo-HSCT, possibly avoiding repeated rounds of consolidation CHT. However, we decided to administer 2 consolidation cycles to our patient, because he achieved suboptimal reduction of molecular MRD after induction. This decision was based on studies showing that NPM1-mutated AML patients MRD-positive immediately before allo-HSCT usually have poor outcome,46-48 although they still do better with allotransplant than with CHT alone,38,58 especially those with >4-log MRD reduction in PB.38 Furthermore, in a recent study,48 NPM1-mutated FLT3-ITD-positive patients with any level of pretransplant MRD positivity showed a 2-year OS of ∼20%. Conversely, patients with pretransplant MRD negativity had a 2-year OS survival approaching 80%, irrespective of the FLT3-ITD status and intensity of transplant conditioning regimen.48 Similar good outcome was also observed in NPM1-mutated FLT3-ITD-negative patients with low levels of pretransplant MRD (ie, <200 copies/105ABL in PB or <1000 copies/105ABL in BM).48 Although reducing pretransplant MRD as much as possible (ideally achieving negativity) in NPM1-mutated FLT3-ITDhigh seems reasonable, the feasibility and clinical significance of this approach remains unknown.
Scenario 3: Old unfit patient with NPM1-mutated AML without FLT3-ITD plus multilineage dysplasia
A 76-year-old man presented with pneumonia and fever. CBC revealed WBC 0.87 × 109/L, Hb 8.0 g/dL, and platelets 121 × 109/L. The BM was infiltrated by AML (blasts 40%) with dysplasia in >50% of myeloid and erythroid progenitors. Megakaryocytes were also dysplastic. Cells of all 3 hematopoietic lineages were cytoplasmic positive for NPM1 (Figure 3A-D). Molecular studies identified a NPM1 mutation, type A and FLT3 wild-type. The BM karyotype was normal. As the patient was unfit for intensive CHT, he was treated with dactinomycin within a clinical trial (EudraCT 2014-003490-41), achieving CR and >3-log reduction in NPM1 transcripts. Ten months later, the patient relapsed. He received off-label venetoclax plus the hypomethylating agent (HMA) 5-azacytidine, achieving CR and marked reduction in NPM1mut transcripts. He is now at his fifth cycle of venetoclax plus HMA.
![Multilineage dysplasia in NPM1-mutated AML. (A) Aberrant cytoplasmic expression of NPM1 in leukemic cells of the 3 hematopoietic cell lineages. (BM biopsy, APAAP staining; original magnification ×400). (B) An area showing several dysplastic megakaryocytes detected by immunostaining against LAT (linker for activation of T cells) (BM biopsy, APAAP staining; original magnification ×400). (C) An area occupied by numerous leukemic cells of erythroid cell lineage that show surface expression of glycophorin (Gly; BM biopsy, APAAP staining; original magnification × 400). (D) Leukemic cells stained for glycophorin (brown) and cytoplasmic NPM1 (blue) (BM biopsy, immunoperoxidase [brown]/APAAP [blue]; original magnification ×400). (E) Diagnostic algorithm for the differential diagnosis between NPM1-mutated AML and AML-MRC. *No prior cytotoxic or radiation therapy; no recurrent cytogenetic abnormalities defining distinct AML entities of 2017 WHO classification.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/5/10.1182_blood.2020008211/1/m_bloodbld2020008211f3.png?Expires=1769102067&Signature=XTq2hEUC1vcQZagAr9TI8hhrcuPONaZgIzfE1nFHkRK3Nkt4h54u-EhviwxT6F22SOYinXLlUkIxCIVMXYt4mnGcWXqoZFN7PIa1SWlUKdRLLWzism72KGAVXf8GohqitL2~l5qR3ektz8TKIu0L1TlK7VsIiJQckn~RnE7Qh5NKwNdYcmReQxD3IA9QDwgJJoxoxhy194v4OXxDNN6QiMXueiA4br-PKamMH9S9~W~qonXVLs8yGm7eGzWf8ycC40-~5YW6TJfrIfRoYBcmk33LQ6sUX5SNCCUTcvUnMPM-MNMER07rULWU~kwsJAWPeAZ7OOrfmr9W18lvdOnV7g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Multilineage dysplasia in NPM1-mutated AML. (A) Aberrant cytoplasmic expression of NPM1 in leukemic cells of the 3 hematopoietic cell lineages. (BM biopsy, APAAP staining; original magnification ×400). (B) An area showing several dysplastic megakaryocytes detected by immunostaining against LAT (linker for activation of T cells) (BM biopsy, APAAP staining; original magnification ×400). (C) An area occupied by numerous leukemic cells of erythroid cell lineage that show surface expression of glycophorin (Gly; BM biopsy, APAAP staining; original magnification × 400). (D) Leukemic cells stained for glycophorin (brown) and cytoplasmic NPM1 (blue) (BM biopsy, immunoperoxidase [brown]/APAAP [blue]; original magnification ×400). (E) Diagnostic algorithm for the differential diagnosis between NPM1-mutated AML and AML-MRC. *No prior cytotoxic or radiation therapy; no recurrent cytogenetic abnormalities defining distinct AML entities of 2017 WHO classification.
Multilineage dysplasia in NPM1-mutated AML. (A) Aberrant cytoplasmic expression of NPM1 in leukemic cells of the 3 hematopoietic cell lineages. (BM biopsy, APAAP staining; original magnification ×400). (B) An area showing several dysplastic megakaryocytes detected by immunostaining against LAT (linker for activation of T cells) (BM biopsy, APAAP staining; original magnification ×400). (C) An area occupied by numerous leukemic cells of erythroid cell lineage that show surface expression of glycophorin (Gly; BM biopsy, APAAP staining; original magnification × 400). (D) Leukemic cells stained for glycophorin (brown) and cytoplasmic NPM1 (blue) (BM biopsy, immunoperoxidase [brown]/APAAP [blue]; original magnification ×400). (E) Diagnostic algorithm for the differential diagnosis between NPM1-mutated AML and AML-MRC. *No prior cytotoxic or radiation therapy; no recurrent cytogenetic abnormalities defining distinct AML entities of 2017 WHO classification.
Questions and recommendations
The low WBC count and morphological features observed in our patient poses the problem of differential diagnosis between NPM1-mutated AML with multilineage dysplasia and AML with myelodisplasia-related changes (AML-MRC). Distinction of these 2 entities is critical, since postremission therapy significantly differs between favorable-risk NPM1-mutated AML with multilineage dysplasia and AML-MRC.60
In our experience, low WBC count is still consistent with a diagnosis of NPM1-mutated AML, especially in cases with wild-type FLT3. According to the 2017 WHO classification, multilineage dysplasia is defined as the presence of dysplasia in at least 2 BM cell lines. This occurs in ∼23% of patients with NPM1-mutated AML.61 In BM biopsies of NPM1-mutated AML patients, megakaryocytes are often increased in number and dysplastic,62 but, unlike AML-MRC or myelodysplastic syndrome (MDS), they are consistently CD34 negative. This is in keeping with our finding that NPM1 mutant A perturbs megakaryopoiesis in a conditional mouse model63 and may also explain the relatively high platelets count in our patient.
The demonstration that NPM1-mutated AML with and without multilineage dysplasia show the same immunophenotype, gene expression profile, and clinical outcome61 led to a major change in the 2017 edition of WHO classification (as compared with the previous 2010 version). Specifically, when NPM1 mutation and multilineage dysplasia coexist, the genetic lesion supersedes morphology and the case is diagnosed as NPM1-mutated AML.8,61 Conversely, a previous history of MDS or the finding of MRC-related cytogenetic abnormalities, even in the presence of NPM1 mutation, are diagnostic of AML-MRC. Our patient was diagnosed as NPM1-mutated AML, since multilineage dysplasia but no history of MDS or specific cytogenetic abnormalities were found. A diagnostic algorithm for distinguishing NPM1-mutated AML with multilineage dysplasia from AML with MRC is shown in Figure 3E.
What is the best therapy available for this patient? Older unfit NPM1-mutated AML patients rarely show durable response to single HMA.64 Thus, combining venetoclax with a HMA (not yet approved by the Italian Drug Agency when we first treated the patient) is now the standard of care, inducing CR in 70% to 90% of NPM1-mutated AML patients65,66 (Figure 4). Most patients achieve CR after 1 cycle. Prophylactic measures should be taken to prevent the venetoclax-induced tumor lysis syndrome, although the risk is lower than in chronic lymphocytic leukemia.67
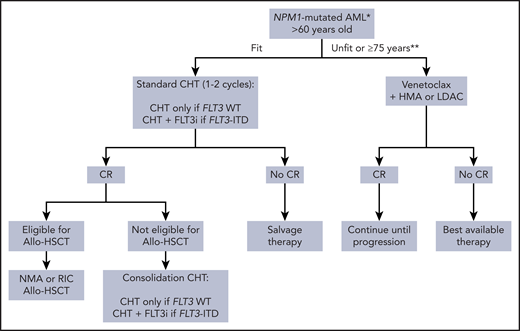
Treatment algorithm for older patients with NPM1-mutated AML. *Include NPM1-mutated AML independently by the FLT3-ITD that does not appear prognostically relevant in older patients.32,74 **Venetoclax-based regimens are approved for fit AML patients ≥75 years by the Italian Drug Agency. During the COVID-19 pandemic, venetoclax-based regimens may be used temporarily in alternative to intensive CHT in older fit NPM1-mutated AML patients,69 to help bridge patients through the pandemic with the aim of subsequently delivering definitive therapy, including allo-HSCT, if required. LDAC, low-dose cytarabine; NMA, nonmyeloablative; RIC, reduced intensity conditioning.
Treatment algorithm for older patients with NPM1-mutated AML. *Include NPM1-mutated AML independently by the FLT3-ITD that does not appear prognostically relevant in older patients.32,74 **Venetoclax-based regimens are approved for fit AML patients ≥75 years by the Italian Drug Agency. During the COVID-19 pandemic, venetoclax-based regimens may be used temporarily in alternative to intensive CHT in older fit NPM1-mutated AML patients,69 to help bridge patients through the pandemic with the aim of subsequently delivering definitive therapy, including allo-HSCT, if required. LDAC, low-dose cytarabine; NMA, nonmyeloablative; RIC, reduced intensity conditioning.
Main side effects of venetoclax plus HMAs are hematological, with ∼40% of cases showing cytopenias and ∼30% grade ≥3 infectious complications.68 These may require dose reductions or interruption of venetoclax. Other common nonhematological adverse events of venetoclax plus 5-azacytidine are gastrointestinal.66 However, mortality at 30 days was only 7% with this combo,66 clearly indicating the safety of this regimen in unfit patients. Venetoclax is a cytochrome P450 3A4 substrate. Thus, its dose should be reduced by at ≥75% when used in combination with posaconazole, a strong cytochrome P450 3A4 inhibitor commonly used for antifungal prophylaxis in AML.67
Venetoclax-based regimens may be also considered as a temporary alternative to intensive CHT in older fit NPM1-mutated AML patients during the COVID-19 pandemic.69,70 Under these circumstances, monitoring of MRD is highly recommended, although the optimal time points for assessment of NPM1mut transcripts level needs yet to be established for venetoclax-based regimens.71
Older fit patients (>60 years) with NPM1-mutated AML should be treated intensively, since they respond better than other genotypes to induction-consolidation CHT72 (Figure 4). However, the outcome remains adverse (15% to 20% OS) independently of the FLT3 status.72,73 In fact, patients >65 years with NPM1-mutated AML without FLT3-ITD exhibit a prognosis poorer than younger patients with the same genotype (2-year OS, 27% vs 70%, respectively).74 Older NPM1-mutated AML patients with FLT3-ITDlow also experience poor outcomes.32 However, this scenario may change rapidly. Improvement in event-free survival has been reported in older AML patients with FLT3-ITD following the addition of midostaurin to CHT.75 Moreover, venetoclax-based regimens have the potential to challenge standard CHT as frontline treatment in NPM1-mutated AML.65 Recently, venetoclax in combination with “5+2” induction CHT (CAVEAT trial) followed by 4 consolidation cycles was found to be safe in fit older patients with AML.76 Deep molecular responses were achieved in most cases of NPM1-mutated AML. The major limiting toxicity in the trial was prolonged thrombocytopenia. This is probably due to the fact that venetoclax may inhibit p-glycoprotein, enhancing intracellular accumulation of idarubicin with consequent toxicity on normal hemopoietic stem cells. NPM1-mutated AML without FLT3-ITD can benefit of reduced intensity conditioning77 or nonmyeloablative78 allo-HSCT (Figure 4).
Scenario 4: NPM1-mutated AML relapsing with therapy-related myelodysplasia lacking NPM1 mutation
A 68-year-old women presented with fatigue, dyspnea and chest pain. CBC showed WBC 3.9 × 109/L, Hb 9.7 g/dL, and platelets 178 × 109/L. The BM was massively infiltrated by blasts with myelomonocytic appearance showing cytoplasmic NPM1 and coexpressing myeloperoxidase and macrophage-restricted CD68, while CD34 was negative. NPM1 mutation A and FLT3 wild-type were detected. BM karyotype was normal. A 7+3 induction regimen resulted into CR with a 4-log reduction of NPM1mut transcripts in BM (Figure 5A). She received 2 cytarabine consolidation cycles, achieving MRD negativity but, after 14 months, developed fever and thrombocytopenia (54 × 109/L). BM examination showed MDS with nuclear expression of NPM1. NPM1mut transcripts were undetectable (Figure 5A), while karyotyping and fluorescence-activated cell sorting revealed del(5q), −7 and monoallelic deletion of TP53. She received 2 cycles of 5-azacytidine (Figure 5A) with platelet recovery; del(5q) and −7 disappeared, while TP53 deletion persisted in 90% of nuclei. Because of high-risk cytogenetics,8 she underwent an haploidentical HSCT (Figure 5A) and is now in cytogenetic CR 2.5 years after the allotransplant.
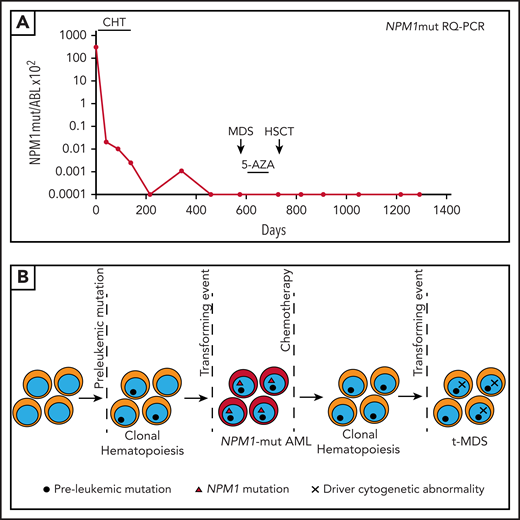
NPM1-mutated AML relapsing as therapy-related MDS. (A) Monitoring of NPM1mut transcripts during therapy and follow-up (see text). 0.0001 is equivalent to MRD negativity. (B) Cartoon depicting the clonal architecture of the BM over time in the patient described in scenario 4. Absence of NPM1 mutations and presence of MRC-related cytogenetic abnormalities at relapse demonstrated the development of a second myeloid neoplasm (t-MDS). 5-AZA, 5-azacitidine; RQ-PCR, RT-qPCR.
NPM1-mutated AML relapsing as therapy-related MDS. (A) Monitoring of NPM1mut transcripts during therapy and follow-up (see text). 0.0001 is equivalent to MRD negativity. (B) Cartoon depicting the clonal architecture of the BM over time in the patient described in scenario 4. Absence of NPM1 mutations and presence of MRC-related cytogenetic abnormalities at relapse demonstrated the development of a second myeloid neoplasm (t-MDS). 5-AZA, 5-azacitidine; RQ-PCR, RT-qPCR.
Questions and recommendations
This case addresses the issue of clonal evolution in NPM1-mutated AML. Approximately 10% of NPM1-mutated AML patients relapse with no detectable NPM1 mutation.79 These cases represent second AML emerging after the eradication of the original NPM1-mutated clone, as proven by different mutational patterns at diagnosis and relapse.80,81 This event may be facilitated by preexisting clonal hematopoiesis and usually occurs after several years.79-81 The second AML may represent either a de novo or therapy-related event (Figure 5B). In our patient, the morphology and fluorescence-activated cell sorting at the time of relapse were diagnostic of therapy-related MDS (t-MDS).8
Except for the short time (∼1 year) before t-MDS development, our case resembles those reported by Herold et al.82 Specifically, 5 patients (median age, 58 years; age range, 47-74 years) initially diagnosed as NPM1-mutated AML developed t-MDS after a median of 46 months (range, 28-92 months) remaining MRD negative for NPM1mut. In 3/5 cases, the karyotype was normal while 2 cases showed, like our patient, chromosome 7 abnormalities.82 Mutational analysis exhibited persistence of clonal hematopoiesis that may have favored the emergence of t-MDS,82 as also reported in another study.83 We had no information on preleukemic clonal hematopoiesis in our patient.
Future perspectives and conclusions
Great progress has been made during the past 15 years in the diagnosis and therapy of NPM1-mutated AML, but there are a number of issues that remain to be clarified and objectives that need to be accomplished.
According to the 2017 WHO classification,8 the percentage of blasts required for diagnosing NPM1-mutated AML must be ≥20%.8 This criterion differs from that adopted for AML with t(8;21), inv(16), t(16;16), and t(15;17), which can be diagnosed regardless of blast cell count. This is due to the report of very rare cases of NPM1-mutated MDS and chronic myelomonocytic leukemia (CMML).84,85 However, NPM1 mutations in CMML associate with normal cytogenetics, “dysplastic phenotype,” DNMT3A mutations,84 higher risk of blastic transformation, and outcomes poorer than CMML without NPM1 mutations.84 Moreover, dramatic monocytosis is sometimes observed in patients with NPM1-mutated AML (especially M5b), in keeping with findings in immunocompromised mice transplanted with CD34+ HSCs from NPM1-mutated AML patients.86 Similarly, cases of NPM1-mutated MDS show features typical of AML, including good response to intensive chemotherapy.85,87 Thus, NPM1 mutations may define AML, irrespective of blast percentage.88 Under these circumstances, we recommend performing a BM biopsy and IHC for cytoplasmic NPM1, since these procedures together allow to evaluate the percentage of blasts better than conventional morphology and immunophenotype. We currently treat patients with NPM1 mutations and <20% of blasts as NPM1-mutated AML, although we recognize that this still represents a controversial issue that requires further studies.
The ELN model is an oversimplification of the real world, since the prognosis of NPM1-mutated AML also depends on comutated genes other than FLT3.89 For example, the NPM1/N-RAS, NPM1/RAD2189 or NPM1/FLT3-tyrosine kinase domain90 genotypes seem to have a relatively favorable outcome, while the NPM1/DNMT3A/FLT3-ITD89 and NPM1/WT191 genotypes show a particularly poor prognosis. Other genotypes occur at very low frequency, making the study of their prognostic impact difficult. It is likely that future versions of 2017 ELN model will be updated according to therapeutic progresses, such as taking into consideration the results achieved with FLT3 inhibitors that were not available when the current ELN risk categorization was established.
MRD evaluation also plays an important role in proper relapse-risk estimation in NPM1-mutated AML. Today, RT-qPCR10 represents the gold standard for assessing levels of NPM1mut transcripts, since it is inexpensive and can be used for MRD monitoring in >90% of NPM1-mutated AML cases. However, sensitivity of next-generation sequencing for MRD monitoring has improved,92-95 and it is expected that this technique will be increasingly used in the future, especially for rare NPM1 mutations. Currently, ELN15 recommends using next-generation sequencing only in clinical trials. Standardizing of NPM1 MRD measurement and better defining its impact at different time points after therapy as well designing NPM1 MRD-based prospective clinical trials is also warranted.
Venetoclax, in combination with CHT, should be further investigated in NPM1-mutated AML. Also, unfit NPM1-mutated AML patients relapsing after venetoclax-based regimens represent a medical need, and further studies should be directed to better understand the mechanisms of resistance to venetoclax. FLT3 mutations (frequent in NPM1-mutated AML) may promote resistance to venetoclax by enhancing expression of other members of the BCL-2 family, including BCL‐XL and MCL‐1.96 Therefore, there is a rationale in using venetoclax plus FLT3 inhibitors in NPM1-mutated AML comutated for FLT3. Phase 1/2 studies combining venetoclax with gilteritinib (NCT03625505) or quizartinib (NCT03735875) are ongoing.
Promising future new agents in NPM1-mutated AML include XPO15 and MLL-Menin97,98 inhibitors, alone or in combination with FLT3 inhibitors99 or venetoclax100 and drugs targeting the interaction between NPM1 and its ligands.101
In conclusion, a better knowledge of the mechanisms underlying leukemic transformation mediated by mutant NPM1 and the design of MRD-focused clinical trials are expected to further improve and personalize the therapy of NPM1-mutated AML patients.
Acknowledgments
This work was supported by the Associazione Italiana per la Ricerca sul Cancro (grants IG 2019 23604 and AIRC Start-Up 2019 22895), the European Research Council (advanced grant 2016 740230 and consolidator grant 2016 725725), and the ARC Foundation for Cancer Research (Leopold Griffuel Prize to B.F.).
Authorship
Contribution: B.F. coordinated the preparation of the review, and all authors cowrote manuscript and were involved in the diagnosis or therapy of the described cases.
Conflict-of-interest disclosure: B.F. licensed a patent on NPM1 mutants (number 102004901256449). B.F. and M.P.M. receives honoraria from Rasna Therapeutics for scientific advisor activities. M.P.M. is a consultant and scientific advisory board member of AbbVie, Amgen, Celgene, Janssen, Novartis, Pfizer, and Jazz Pharmaceuticals and receives honoraria from Amgen, Celgene, Janssen, and Novartis. L.B. declares consultancy at scientific advisory boards for AbbVie.
Correspondence: Brunangelo Falini, Institute of Hematology and CREO, Piazzale Menghini 9, University of Perugia, 06131 Perugia, Italy; e-mail: brunangelo.falini@unipg.it.
