

Key Points
Strong classical pathway activation leads to C5 activation despite C3 inhibition signifying C3 bypass activation of C5.
Conformational activation of C5 in absence of convertases or other enzymes cannot be inhibited by different individual C5 inhibitors.

Abstract
Blocking the terminal complement pathway with the C5 inhibitor eculizumab has revolutionized the clinical management of several complement-mediated diseases and has boosted the clinical development of new inhibitors. Data on the C3 inhibitor Compstatin and the C5 inhibitors eculizumab and Coversin reported here demonstrate that C3/C5 convertases function differently from prevailing concepts. Stoichiometric C3 inhibition failed to inhibit C5 activation and lytic activity during strong classical pathway activation, demonstrating a “C3 bypass” activation of C5. We show that, instead of C3b, surface-deposited C4b alone can also recruit and prime C5 for consecutive proteolytic activation. Surface-bound C3b and C4b possess similar affinities for C5. By demonstrating that the fluid phase convertase C3bBb is sufficient to cleave C5 as long as C5 is bound on C3b/C4b-decorated surfaces, we show that surface fixation is necessary only for the C3b/C4b opsonins that prime C5 but not for the catalytic convertase unit C3bBb. Of note, at very high C3b densities, we observed membrane attack complex formation in absence of C5-activating enzymes. This is explained by a conformational activation in which C5 adopts a C5b-like conformation when bound to densely C3b-opsonized surfaces. Stoichiometric C5 inhibitors failed to prevent conformational C5 activation, which explains the clinical phenomenon of residual C5 activity documented for different inhibitors of C5. The new insights into the mechanism of C3/C5 convertases provided here have important implications for the development and therapeutic use of complement inhibitors as well as the interpretation of former clinical and preclinical data.
Introduction
The initiation of the complement terminal pathway (TP) by proteolytic activation of C5 has received enormous interest since the anti-C5 antibody eculizumab has been approved for therapeutic use in the diseases paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome (aHUS), anti-acetylcholine receptor antibody positive generalized myasthenia gravis, and anti-aquaporin-4 antibody-positive neuromyelitis optica spectrum disorder.1-4 Recently, a second-generation version of eculizumab with improved pharmacokinetic properties, ravulizumab, has entered the clinic.5,6 Both monoclonal antibodies bind C5 with picomolar affinity and prevent its proteolytic activation into the anaphylatoxin C5a and C5b, which assembles the membrane attack complex (MAC).7,8 Given the therapeutic success of eculizumab, complement intervention at the level of C5 is currently the most sought-after strategy in the developmental pipeline. More than 10 different C5 inhibiting approaches are currently being investigated in clinical trials for several different clinical indications.9
And yet C5 inhibition in the clinic can have its pitfalls. For some aHUS patients TP activation markers in eculizumab-treated patients were detected consistent with eculizumab not completely blocking TP activity in cell culture experiments.10,11 In PNH patients, extravascular hemolysis and continuous subclinical breakthrough hemolysis have been described during eculizumab therapy.12,13 Even during apparently adequate levels of eculizumab/ravulizumab, hemolysis can reoccur in PNH patients when they are exposed to complement-amplifying conditions such as infection and surgery.14,15 The same issues were described to occur during anti-C5 therapy with Coversin.16 Strong proximal complement activity in such conditions can lead to a highly dense cellular C3b deposition that correlates with residual TP activity and cell lysis.17 Such incidences in the clinic have been described as pharmacodynamic breakthrough,15,18,19 but the underlying molecular mechanism remains elusive, impeding predictions on why, when, and to what extent pharmacodynamic breakthrough can occur.
To overcome the sometimes-suboptimal response to anti-C5 therapy, new therapeutic strategies have been developed. Prominent examples are peptide inhibitors of the Compstatin family that bind C3 with high affinity and block its activation by convertases. Four members of the Compstatin family have been evaluated for their therapeutic potential in clinical trials. Although most indications trialed are alternative pathway (AP)-mediated diseases like PNH or C3 glomerulopathy; certain Compstatin analogs have also been investigated in diseases mediated by other complement pathways (classical pathway [CP] and/or lectin pathway [LP]) (ie, ABO-incompatible kidney transplantation, autoimmune hemolytic anemia, and COVID-19).9,20 For these scenarios, it is crucial to know whether the C3 inhibition approaches are able to completely protect from C5 activation by the CP. Indeed, here we demonstrate that stoichiometric C3 inhibition is associated with remaining TP activity and cell lysis.
Taken together, different clinically developed or therapeutically used anti-complement agents that target the complement cascade at the levels of C3 or C5 fail to reliably prevent TP activation. We hypothesized that the complement cascade runs differently than expected under conditions when the pathways are intercepted pharmacologically at critical steps. To prove this and uncover the underlying mechanism was the aim of this study. As a result, this study informs on when and why C3 and C5 inhibitors will fail to completely inhibit the lytic complement pathway and provides fundamental new insights into complement biology.
Methods
Blood components
Sera from healthy donors were collected in VACUETTE/S-Monovette serum collection tubes, aliquoted, frozen in liquid nitrogen, and stored at −80°C. Blood from healthy volunteers was used with approval by the Ulm University Ethics Committee. Rabbit and sheep erythrocytes in Alsever’s solution were obtained from Fiebig Nährstofftechnik GbR (Idstein, Germany) and stored at 4°C. Factor I-depleted, C3- and C5-depleted, and normal human serum (NHS) were obtained from CompTech. Other reagents are detailed in the supplemental Methods, available on the Blood Web site.
Assays with erythrocytes
C3b- and C4b-opsonization and CP/AP hemolysis assays including the respective detection by flow cytometry or determining absorbance values were performed as described previously, partly with variations.17 Details are given in the supplemental Methods.
Cell culture
Human umbilical vein endothelial cells were grown in endothelial growth medium MV2 (ready-to-use, C-22022; PromoCell) at 37°C and 5% carbon dioxide and exposed to CP as detailed in the supplemental Methods.
Multiplex analysis of serum samples
Selected serum samples were assayed for different chemokines/cytokines by using Multiplex analysis (MAP Human Cytokine/Chemokine Magnetic Bead Panel; Milliplex).
Generation of C5conf-6
C5conf-6 formation was performed on the basis of previous work.21 A total of 25 µL C5 was mixed with 12.5 µL C6 and run through 30 freeze/thaw cycles. Mixtures were aliquoted and stored at −80°C until further use. For generation of C5conf-9, 1 µL C7 (CompTech), 2 µL C8 (CompTech), and 2 µL C9 (CompTech) were added to 5 µL of C5conf-6 and incubated for 15 minutes at 37°C. Stocks of the proteins C5 to C9 were at 1 mg/ml. Detection was performed by enzyme-linked immunosorbent assay as detailed in the supplemental Methods.
SPR
Determination of affinities and analytical protein–protein interactions were performed basically by using the same methods and equipment as previously described.17,22,23
Statistical analysis
The statistical analyses were performed using the software GraphPad Prism (version 8.0.2). The details of the statistical tests are given in the respective figure legends. Values of P < .05 were considered to be statistically significant for all tests and significance is denoted by an asterisk.
Detailed descriptions of assay steps can be found in the supplemental Methods.
Results
C3 inhibition fails to block CP mediated C5 activation
The Compstatin version Cp40 was applied to inhibit CP and AP-mediated TP activity. Although AP-mediated lysis of rabbit erythrocytes (rabbit red blood cells [rRBCs]) was entirely inhibited by Cp40, the peptide inhibitor allowed considerable CP-mediated hemolysis of sensitized sheep erythrocytes (sensitized sheep red blood cells [shRBCs]) in conditions containing 80% NHS (Figure 1A). In an ABO-incompatible human whole blood model, eculizumab and Cp40 dramatically reduced but could not completely prevent CP-mediated lysis (Figure 1B). The observed levels of RBC lysis varied according to the donor blood used, but the effect size within each donor was also variable because of different levels of blood clot formation that sometimes occurred during the assay. Nevertheless, these data demonstrate that C3 and C5 inhibitors do not reliably prevent lysis of healthy human RBCs after strong CP activation. Recently, Zhang et al reported that absence of C3 in serum does not prevent CP-mediated hemolysis in cases when serum percentages are higher than 5%.24 Indeed, commercially available C3-depleted serum efficiently lysed sensitized shRBCs (supplemental Figure 1A). Serum percentage also influences the level of CP-mediated hemolysis in presence of excess amounts of a stoichiometric C3 inhibitor. A threefold molar excess of Cp40 over C3 in all serum percentages assayed could not completely inhibit hemolysis. The level of lysis depended not only on the serum percentage, but also on the antibody used for sensitization. Sensitization with rabbit anti-shRBC antiserum yielded higher hemolysis level in Cp40-inhibited NHS rather than NHS containing naturally occurring antibodies against the Forssman antigen on shRBCs (Figure 1C-D). Hemolysis under C3 inhibition was complement dependent because double C5 inhibition or C5-depleted serum protected from lysis (supplemental Figure 1B-C). Very strong CP activation in presence of C3 inhibition leads to cell activation of nucleated cells, yielding increased cytokine secretion by cultured endothelial cells (supplemental Figure 1D-F). Increased cytokine release depended on C5 activation because inhibition of C5 protected from increased cytokine production. Having confirmed that C5 activation after CP activation is indeed possible in the absence of C3 or in NHS with excess of Cp40, the important question remains whether Cp40 can completely inhibit C3 deposition. Therefore, we investigated the surface opsonization of the cells after CP and AP activation. Hemolysis assays were performed as previously but in the presence of C5 double inhibition with eculizumab and Coversin to completely prevent cell lysis.15,17 Having activated the CP, we observed highest C3 and C4 deposition levels (fluorescence signals >104) when no inhibitor was present. In the presence of Cp40, C4 deposition was high, as expected, but, remarkably, C3 deposition was substantially reduced but not completely absent (Figure 1E). For comparison, we also tested C3 and C4 surface deposition after activating the AP while C3 was inhibited (Figure 1F). Under these conditions, neither C3 nor C4 fragments were deposited on the cell surfaces, indicating that Cp40 completely inhibits AP but not CP mediated C3 activation by the respective convertases.
![Strong CP activation leads to hemolysis despite of C3 peptide inhibitor Cp40. (A) CP- and AP-mediated lysis in presence of Cp40. A total of 80% NHS naturally containing antibodies against the Forssman antigen (which is present on shRBCs) was mixed with Cp40 to obtain final Cp40 concentrations of 0.25 to 16 µM, as indicated. AP activity was determined by mixing rabbit erythrocytes with 80% NHS supplemented with 5 mM Mg-EGTA in the presence of the same range of Cp40 concentrations. A serum mix with phosphate-buffered saline (PBS) instead of Cp40 served as positive control, whereas 5 mM EDTA and 5 mM Mg-EGTA dissolved in PBS (in case of CP; for each final concentration) served as negative controls (PBSE, PBS containing EDTA). Released hemoglobin was measured as a marker of hemolysis (average of 3 independent assays with standard deviation [SD] is shown). (B) ABO-incompatible human whole blood model. Human RBCs of blood group AB was added into human whole blood from 2 different donors with blood group O and incubated at 37°C for 3 hours in a reaction tube that had been surface treated with an anticoagulant. Absorbance of released hemoglobin was measured (after blank subtraction) at 415 nm as a marker of hemolysis. Final concentrations of analytes in the assay were: EDTA 10 mM (PBSE), eculizumab 1 µM (Ecu), Cp40 15 µM. The average of 6 independent assays with SD is shown. The observed absolute Abs (415 nm) ranges, after being corrected for the blank values, were for donor 1: EDTA, −0.01 to 0.17; Ecu, 0.03-0.55; Cp40, 0.06-0.60; for donor 2: EDTA, −0.03 to 0.07; Ecu, 0-0.19; Cp40: 0-0.17). An ordinary 1-way analysis of variance (ANOVA) using Tukey's multiple comparisons test was applied ns, not significant. (C) Forssman antigen induced activation of CP in presence of Cp40 at varying serum concentrations. Hemolysin-sensitized shRBCs were incubated in varying serum (containing antibodies against Forssman antigen) concentrations from 1.25% to 80%. Cp40 concentration in 80% serum was 16 µM and diluted accordingly, together with the serum resulting in a constant threefold molar excess of Cp40 over C3 in the serum. PBS, 5 mM EDTA, or Mg-EGTA served as controls as in panel A. Released hemoglobin was measured as a marker of hemolysis (average of 3 independent assays with SD is shown). (D) As in panel B, with the difference that shRBCs were sensitized with hemolysin before being exposed to NHS from which antibodies against the Forssman antigen had been preabsorbed. (E) C3/C4 surface opsonization after CP activation. shRBCs were assayed in 80% serum containing antibodies against the Forssman antigen in presence of C5 double inhibition (eculizumab and Coversin each at a final concentration of 0.5 µM to completely inhibit hemolysis). Then, cells were stained with an anti-human C3c mAb and C4c mAb. Fluorescence signals (median fluorescent index [MFI]) were measured by flow cytometry (average of 3 independent assays with SD is shown). (F) C3/C4 surface opsonization after AP activation. rRBCs were mixed with 80% preabsorbed NHS supplemented with 5 mM Mg-EGTA in presence of double C5 inhibition (by eculizumab and Coversin each at a final concentration of 0.5 µM) and the specified analytes. Cells were stained with an anti-human C3c mAb and C4c mAb. Detection and data presentation as in panel D.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/4/10.1182_blood.2020005959/1/m_bloodbld2020005959f1.png?Expires=1771331891&Signature=hSuKUoqfVWctEM3RYuXRcvH218icfxLc6S2TblOxsCtFwA2pXVIBrvMq-oOh2JjG6gHC-3UimbnNO9VrVmxCmc4Rk1hQMQx8kdPN13Vuzopf1J~w~kdKk0L3dq~35xRALrE1DCwJNiRMvtDwOjyz2hs6AbcwDarRspYPTNrLk3ZvDRc-rbk7gYZSm4A283T1CoOBP5Pg7cIh5A0ic4NGE4shc1-R5RWNGU9Uk4n4PiFZc6d6w94YbwBHwKcUWL-9PkrZYJulpqMINWcx5n97AcW2j2gjYEO9WGtuboXUqi4s-ZIfQ9NUNZ-qGGgQVLet50CHmn2XYl1GZjskWMCBwg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Strong CP activation leads to hemolysis despite of C3 peptide inhibitor Cp40. (A) CP- and AP-mediated lysis in presence of Cp40. A total of 80% NHS naturally containing antibodies against the Forssman antigen (which is present on shRBCs) was mixed with Cp40 to obtain final Cp40 concentrations of 0.25 to 16 µM, as indicated. AP activity was determined by mixing rabbit erythrocytes with 80% NHS supplemented with 5 mM Mg-EGTA in the presence of the same range of Cp40 concentrations. A serum mix with phosphate-buffered saline (PBS) instead of Cp40 served as positive control, whereas 5 mM EDTA and 5 mM Mg-EGTA dissolved in PBS (in case of CP; for each final concentration) served as negative controls (PBSE, PBS containing EDTA). Released hemoglobin was measured as a marker of hemolysis (average of 3 independent assays with standard deviation [SD] is shown). (B) ABO-incompatible human whole blood model. Human RBCs of blood group AB was added into human whole blood from 2 different donors with blood group O and incubated at 37°C for 3 hours in a reaction tube that had been surface treated with an anticoagulant. Absorbance of released hemoglobin was measured (after blank subtraction) at 415 nm as a marker of hemolysis. Final concentrations of analytes in the assay were: EDTA 10 mM (PBSE), eculizumab 1 µM (Ecu), Cp40 15 µM. The average of 6 independent assays with SD is shown. The observed absolute Abs (415 nm) ranges, after being corrected for the blank values, were for donor 1: EDTA, −0.01 to 0.17; Ecu, 0.03-0.55; Cp40, 0.06-0.60; for donor 2: EDTA, −0.03 to 0.07; Ecu, 0-0.19; Cp40: 0-0.17). An ordinary 1-way analysis of variance (ANOVA) using Tukey's multiple comparisons test was applied ns, not significant. (C) Forssman antigen induced activation of CP in presence of Cp40 at varying serum concentrations. Hemolysin-sensitized shRBCs were incubated in varying serum (containing antibodies against Forssman antigen) concentrations from 1.25% to 80%. Cp40 concentration in 80% serum was 16 µM and diluted accordingly, together with the serum resulting in a constant threefold molar excess of Cp40 over C3 in the serum. PBS, 5 mM EDTA, or Mg-EGTA served as controls as in panel A. Released hemoglobin was measured as a marker of hemolysis (average of 3 independent assays with SD is shown). (D) As in panel B, with the difference that shRBCs were sensitized with hemolysin before being exposed to NHS from which antibodies against the Forssman antigen had been preabsorbed. (E) C3/C4 surface opsonization after CP activation. shRBCs were assayed in 80% serum containing antibodies against the Forssman antigen in presence of C5 double inhibition (eculizumab and Coversin each at a final concentration of 0.5 µM to completely inhibit hemolysis). Then, cells were stained with an anti-human C3c mAb and C4c mAb. Fluorescence signals (median fluorescent index [MFI]) were measured by flow cytometry (average of 3 independent assays with SD is shown). (F) C3/C4 surface opsonization after AP activation. rRBCs were mixed with 80% preabsorbed NHS supplemented with 5 mM Mg-EGTA in presence of double C5 inhibition (by eculizumab and Coversin each at a final concentration of 0.5 µM) and the specified analytes. Cells were stained with an anti-human C3c mAb and C4c mAb. Detection and data presentation as in panel D.
Strong CP activation leads to hemolysis despite of C3 peptide inhibitor Cp40. (A) CP- and AP-mediated lysis in presence of Cp40. A total of 80% NHS naturally containing antibodies against the Forssman antigen (which is present on shRBCs) was mixed with Cp40 to obtain final Cp40 concentrations of 0.25 to 16 µM, as indicated. AP activity was determined by mixing rabbit erythrocytes with 80% NHS supplemented with 5 mM Mg-EGTA in the presence of the same range of Cp40 concentrations. A serum mix with phosphate-buffered saline (PBS) instead of Cp40 served as positive control, whereas 5 mM EDTA and 5 mM Mg-EGTA dissolved in PBS (in case of CP; for each final concentration) served as negative controls (PBSE, PBS containing EDTA). Released hemoglobin was measured as a marker of hemolysis (average of 3 independent assays with standard deviation [SD] is shown). (B) ABO-incompatible human whole blood model. Human RBCs of blood group AB was added into human whole blood from 2 different donors with blood group O and incubated at 37°C for 3 hours in a reaction tube that had been surface treated with an anticoagulant. Absorbance of released hemoglobin was measured (after blank subtraction) at 415 nm as a marker of hemolysis. Final concentrations of analytes in the assay were: EDTA 10 mM (PBSE), eculizumab 1 µM (Ecu), Cp40 15 µM. The average of 6 independent assays with SD is shown. The observed absolute Abs (415 nm) ranges, after being corrected for the blank values, were for donor 1: EDTA, −0.01 to 0.17; Ecu, 0.03-0.55; Cp40, 0.06-0.60; for donor 2: EDTA, −0.03 to 0.07; Ecu, 0-0.19; Cp40: 0-0.17). An ordinary 1-way analysis of variance (ANOVA) using Tukey's multiple comparisons test was applied ns, not significant. (C) Forssman antigen induced activation of CP in presence of Cp40 at varying serum concentrations. Hemolysin-sensitized shRBCs were incubated in varying serum (containing antibodies against Forssman antigen) concentrations from 1.25% to 80%. Cp40 concentration in 80% serum was 16 µM and diluted accordingly, together with the serum resulting in a constant threefold molar excess of Cp40 over C3 in the serum. PBS, 5 mM EDTA, or Mg-EGTA served as controls as in panel A. Released hemoglobin was measured as a marker of hemolysis (average of 3 independent assays with SD is shown). (D) As in panel B, with the difference that shRBCs were sensitized with hemolysin before being exposed to NHS from which antibodies against the Forssman antigen had been preabsorbed. (E) C3/C4 surface opsonization after CP activation. shRBCs were assayed in 80% serum containing antibodies against the Forssman antigen in presence of C5 double inhibition (eculizumab and Coversin each at a final concentration of 0.5 µM to completely inhibit hemolysis). Then, cells were stained with an anti-human C3c mAb and C4c mAb. Fluorescence signals (median fluorescent index [MFI]) were measured by flow cytometry (average of 3 independent assays with SD is shown). (F) C3/C4 surface opsonization after AP activation. rRBCs were mixed with 80% preabsorbed NHS supplemented with 5 mM Mg-EGTA in presence of double C5 inhibition (by eculizumab and Coversin each at a final concentration of 0.5 µM) and the specified analytes. Cells were stained with an anti-human C3c mAb and C4c mAb. Detection and data presentation as in panel D.
C3b and C4b bind C5 with similar affinity
The prevalent concept on convertase-mediated activation of C5 describes the (additional) generation of C3b molecules as a requirement for shifting the substrate specificity of the convertases C3bBb and C4b2a from C3 to C5.25-28 Different studies have shown a strong correlation between C3b opsonization levels and the activation of C5, introducing a refined concept of C5 activation by which the additional C3b molecules serve as a binding platform for C5 to induce slight conformational changes in C5 that allow proteolytic processing (denominated “C5 priming”).17,29-31 Here, the mechanistic question arises how C5 activation is possible in absence of C3. Therefore, we tested our hypothesis that C5 priming can be performed by deposited C4b as well as C3b. For better comparison, we first estimated binding affinities of C3 and C5 binding to C3b (Figure 2A,B), which were 1.5 µM and 2.5 µM, respectively, establishing that binding of C3 and C5 to the opsonin C3b is feasible without the catalytic subunit Bb. Then we demonstrate that C4b features similar characteristics as C3b, as hypothesized. C3 and C5 binding to C4b occurred in a similar manner and resulted in comparable estimated binding affinities: 5.8 µM for C3 and 1.2 µM for C5 (Figure 2C-D). Because we show that C3 binds directly to C4b, we evaluated if Cp40 can completely inhibit C3 binding to C4b. Although Cp40 almost completely inhibited C3 binding to C3b when both, C3 and C3b were bound by Cp40, the peptide inhibitor only reduced C3 binding to C4b to about 40% (Figure 2E-F).32
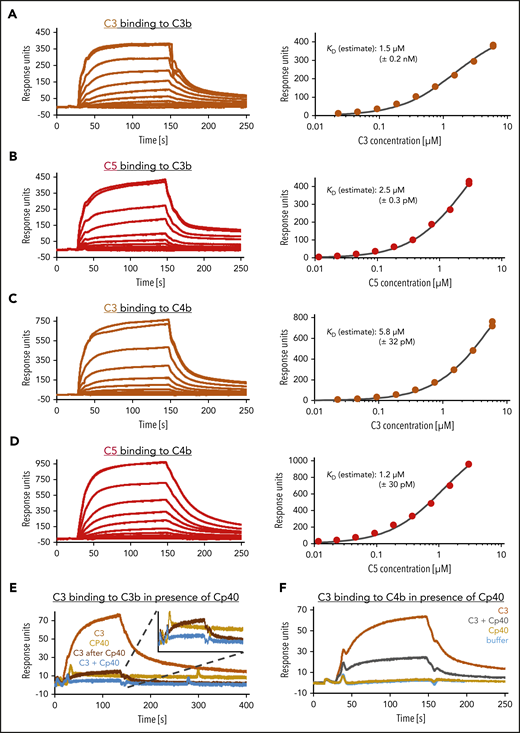
SPR analysis of C3 and C5 binding to C3b and C4b. (A-D) Estimation of equilibrium dissociation constants for C3 and C5 binding to C3b and C4b. After biotinylation of C3b and C4b molecules at their thioester moiety, 1500 and 2100 response units (RUs), respectively, were immobilized onto a streptavidin SAP sensor chip. C3 was flowed over in concentrations ranging from 0.023 to 6 µM, C5 in concentrations from 0.011 to 3 µM. The equilibrium dissociation constant KD was estimated by plotting the steady-state response over the corresponding concentration and using a 1:1 steady-state affinity model. (E) C3 binding to C3b in presence of Cp40. A total of 1800 RUs of biotinylated C3b was immobilized on a SAP sensor chip. C3, at a concentration of 0.6 µM, was applied to the chip (orange) before or (brown) after the immobilized C3b had been exposed to Cp40. The binding of Cp40 (6 µM) to C3b was also assayed (yellow). Finally, a C3 (0.6 µM) together with a 10-fold excess of Cp40 was flowed over the Cp40-saturated C3b surface (blue). (F) C3 binding to C4b despite Cp40 excess. A total of 1700 RUs of biotinylated C4b was immobilized on a SAP sensor chip. C3, at a concentration of 0.35 µM, was flowed over the chip alone (orange) or in mixture with Cp40 at a concentration 10-fold over C3 (gray). Cp40 alone did not bind (yellow). The number of independent SPR assays performed were: 1 typical assay of 2 independent assays (A-B,F); 1 assay was performed (C-D,E).
SPR analysis of C3 and C5 binding to C3b and C4b. (A-D) Estimation of equilibrium dissociation constants for C3 and C5 binding to C3b and C4b. After biotinylation of C3b and C4b molecules at their thioester moiety, 1500 and 2100 response units (RUs), respectively, were immobilized onto a streptavidin SAP sensor chip. C3 was flowed over in concentrations ranging from 0.023 to 6 µM, C5 in concentrations from 0.011 to 3 µM. The equilibrium dissociation constant KD was estimated by plotting the steady-state response over the corresponding concentration and using a 1:1 steady-state affinity model. (E) C3 binding to C3b in presence of Cp40. A total of 1800 RUs of biotinylated C3b was immobilized on a SAP sensor chip. C3, at a concentration of 0.6 µM, was applied to the chip (orange) before or (brown) after the immobilized C3b had been exposed to Cp40. The binding of Cp40 (6 µM) to C3b was also assayed (yellow). Finally, a C3 (0.6 µM) together with a 10-fold excess of Cp40 was flowed over the Cp40-saturated C3b surface (blue). (F) C3 binding to C4b despite Cp40 excess. A total of 1700 RUs of biotinylated C4b was immobilized on a SAP sensor chip. C3, at a concentration of 0.35 µM, was flowed over the chip alone (orange) or in mixture with Cp40 at a concentration 10-fold over C3 (gray). Cp40 alone did not bind (yellow). The number of independent SPR assays performed were: 1 typical assay of 2 independent assays (A-B,F); 1 assay was performed (C-D,E).
Additional binding sites for C5 on C3b
C3 conversion takes place in the fluid phase and on surfaces (even at low C3b deposition), whereas efficient C5 conversion happens on surfaces with a dense deposition of C3b as Berends et al have shown.30,33 Zwarthoff et al concluded that C5 interacts with C3b at the MG4-MG5 site on C3b similarly to what is proposed for C3.32-34 However, contrary to C3, additional sites on C3b were proposed to be important for C5 binding.33 We investigated a second C5 interaction site on C3b different from the MG4-MG5 site. The variable domain of the complement receptor CRIg efficiently blocks the MG4-MG5 site to reduce C3 or C5 binding to C3b.35 Thus, we expressed this domain at high purity to use it in surface plasmon resonance (SPR) competition assays (supplemental Figure 2). When C3, C5, FH19-20, and CRIg were probed individually for binding to immobilized C3b, all proteins bound as expected (Figure 3A; supplemental Figure 3A). The simultaneous injection of 2 C3b ligands (ie, FH19-20 and CRIg, which are known to address different binding sites on C3b) yielded a binding response that equals the sum of the 2 individual injections (supplemental Figure 3A). The same is true for simultaneous injections of FH19-20 and C3/C5, indicating that these molecules do not share overlapping binding patches on C3b (supplemental Figure 3B). The competition assays with CRIg and C3/C5 for C3b binding confirmed that blockage of C3b by CRIg prevents C3 binding, whereas C5 binding is only reduced indicating additional binding sites (Figure 3A). To further test this, we estimated the equilibrium dissociation constants of C3/C5 binding for iC3b. In iC3b, the CUB domain, which links the C3b thioester domain to the body of MG domains, becomes proteolytically processed and consequently partially unfolded, whereas the MG4-MG5 interface remains intact. Hence, the estimated KD for C3 binding to C3b or iC3b, using solely the MG4-MG5 interface, changed only moderately from 1.5 to 2.8 µM. In contrast, C5 bound several-fold weaker to iC3b (KD range, 5-11 µM) than to C3b (KD ∼2.5 µM) (Figure 3B-C). We additionally confirmed that during the processing of C3b into iC3b, the additional interaction site for C5 is lost: CRIg is sufficient to completely block C3 as well as C5 binding when iC3b is the binding partner instead of C3b. This implies that the additional C3b binding site for C5 is either localized directly at the C3b CUB domain or at C3b portions that are spatially oriented by an uncleaved CUB domain. The additional C5 binding site on C3b also becomes impaired upon convertase formation. C3bBb convertase formation weakens the overall binding of C5 to C3b (supplemental Figure 4A-D), which is consistent to what was shown before.17 This further suggests that areas far apart from the C3b MG4-MG5 interface participate in C5 recruitment.
![Identification of a second binding site for C5 at the CUB domain of C3b. (A) Binding competition between C3/C5 and CRIg on C3b. A total of 1500 RUs of biotinylated C3b molecules was immobilized on a streptavidin-coated SAP sensor chip. C3 (0.3 µM) and C5 (0.15 µM) were flowed over the chip alone (brown and red, respectively) or in a mixture with CRIg (30 µM) (purple and blue). The individual injection of CRIg is shown in green. Duplicates of each binding event were performed to show reproducibility. (B-C) Determination of dissociation constants for C3/C5 binding to iC3b. An iC3b immobilization signal of 1100 RUs was reached. C3 was flowed over in concentrations ranging from 0.046 to 6 µM, C5 in concentrations from 0.023 to 3 µM. The equilibrium dissociation constant KD was estimated by plotting the steady-state response over the corresponding concentration and using a 1:1 steady-state affinity model. Note that the estimated KD for the interaction of C5 with iC3b is higher than the highest concentration assayed and thus is less precise when compared with the other KD estimations. (D) As in panel A, but on iC3b. A total of 1100 RUs of biotinylated iC3b was immobilized on a SAP sensor chip. The number of independent SPR assays performed were: 1 typical assay of 2 independent assays (A,C-D [right side, C5 as analyte]); 1 assay was performed (B,D [left side, C3 as analyte]).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/4/10.1182_blood.2020005959/1/m_bloodbld2020005959f3.png?Expires=1771331891&Signature=3t7ziv9eOhB2Zl3azLuYaHwWCFh3gWQx0qB9l8HPPriUQ5FAAXNbnmL36lAX0TQ8CphtQ-FMnAxF8bHbLvCF3pAYfQSH2qTuVbv2bwSqD4AWoLlAPguAZsJSw32yw9CCZQq3p0Bqf1P09rjVO471gcF7dIHxMmCAsF5euPdR~dLV-MTDXJI3RmFYRKoRGGrQzo-Mqr3DNOMZaNZnv30Bys3sgZCksD3HVO4W7-Dsfqs9hNWgclzgLEBLufm2yjn4ml-S0sP~vBTGFEh7ThBPnx1s74Au3t37evwcPmljwDvuaWmMTypabgQZWNE~1rxh0NmwyyU410QpqV388c3NKQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Identification of a second binding site for C5 at the CUB domain of C3b. (A) Binding competition between C3/C5 and CRIg on C3b. A total of 1500 RUs of biotinylated C3b molecules was immobilized on a streptavidin-coated SAP sensor chip. C3 (0.3 µM) and C5 (0.15 µM) were flowed over the chip alone (brown and red, respectively) or in a mixture with CRIg (30 µM) (purple and blue). The individual injection of CRIg is shown in green. Duplicates of each binding event were performed to show reproducibility. (B-C) Determination of dissociation constants for C3/C5 binding to iC3b. An iC3b immobilization signal of 1100 RUs was reached. C3 was flowed over in concentrations ranging from 0.046 to 6 µM, C5 in concentrations from 0.023 to 3 µM. The equilibrium dissociation constant KD was estimated by plotting the steady-state response over the corresponding concentration and using a 1:1 steady-state affinity model. Note that the estimated KD for the interaction of C5 with iC3b is higher than the highest concentration assayed and thus is less precise when compared with the other KD estimations. (D) As in panel A, but on iC3b. A total of 1100 RUs of biotinylated iC3b was immobilized on a SAP sensor chip. The number of independent SPR assays performed were: 1 typical assay of 2 independent assays (A,C-D [right side, C5 as analyte]); 1 assay was performed (B,D [left side, C3 as analyte]).
Identification of a second binding site for C5 at the CUB domain of C3b. (A) Binding competition between C3/C5 and CRIg on C3b. A total of 1500 RUs of biotinylated C3b molecules was immobilized on a streptavidin-coated SAP sensor chip. C3 (0.3 µM) and C5 (0.15 µM) were flowed over the chip alone (brown and red, respectively) or in a mixture with CRIg (30 µM) (purple and blue). The individual injection of CRIg is shown in green. Duplicates of each binding event were performed to show reproducibility. (B-C) Determination of dissociation constants for C3/C5 binding to iC3b. An iC3b immobilization signal of 1100 RUs was reached. C3 was flowed over in concentrations ranging from 0.046 to 6 µM, C5 in concentrations from 0.023 to 3 µM. The equilibrium dissociation constant KD was estimated by plotting the steady-state response over the corresponding concentration and using a 1:1 steady-state affinity model. Note that the estimated KD for the interaction of C5 with iC3b is higher than the highest concentration assayed and thus is less precise when compared with the other KD estimations. (D) As in panel A, but on iC3b. A total of 1100 RUs of biotinylated iC3b was immobilized on a SAP sensor chip. The number of independent SPR assays performed were: 1 typical assay of 2 independent assays (A,C-D [right side, C5 as analyte]); 1 assay was performed (B,D [left side, C3 as analyte]).
C5 priming by C3b or C4b as prerequisite for enzymatic and nonenzymatic C5 activation
Remarkably, the presence of the enzymatic unit Bb on C3b reduces the recruitment of C5 to C3b (supplemental Figure 4A-D). Hence, we hypothesized that the additional C3b molecules necessary for efficient C5 activation do not associate with C3bBb but rather independently bind and alter the conformation of captured C5 to facilitate its subsequent cleavage by the bimolecular convertase C3bBb. Indeed, C3b is neither able to bind to immobilized C3b nor to the bimolecular convertase C3bBb, questioning the existence of the trimolecular C5 convertase C3bBb3b (Figure 4A-B). This raises the question whether the bimolecular C3 convertase C3bBb is also the C5 convertase with the important prerequisite that C5 can only be enzymatically processed when it is prepared for cleavage (or primed) on a densely C3b or C4b opsonized surface as has already been proposed before in the case of C3b opsonins.29 Therefore, we either mixed untreated or C3b preopsonized rRBCs with the purified complement components C5, C6, C7, C8, and C9 and exposed these mixtures to bimolecular fluid phase convertases C3bBb (Figure 4C-D). Importantly, in absence of decay accelerators, the assembled C3bBb convertases can remain active for several minutes as presented in supplemental Figure 5A and shown before.36 Although nonopsonized rRBCs lysed only in the water control, C3b opsonized rRBCs lysed also when the complement components C5-9 were added in presence and, remarkably, also in absence of fluid phase convertases C3bBb. The C3b preopsonized rRBC had been incubated several times with recombinant complement decay accelerating proteins to ensure efficient convertases decay after the C3b opsonization step prior usage in the assay. The sole addition of C5-9 to C3b preopsonized rRBCs in absence of convertases led to lysis of ∼71%. We hypothesized that prolonged priming on highly dense C3b surfaces leads to a conformational activation of C5 which consecutively recruits C6-9 to form pores for cell lysis. To prove that C5 activation is the initiating event in this process, preopsonized rRBCs were also exposed to C6-9 (supplemental Figure 5B) but no lysis was observed then. To rule out that any convertase activity was involuntarily introduced through potential low-level contaminations with factor B and factor D within the purified components C5-9, the same experiment was repeated in EDTA buffer because absence of Mg ions prevents convertase assembly (supplemental Figure 5C). However, the activity of priming or conformationally activating C5 on densely or very densely opsonized surfaces, respectively, is not unique for C3b. Surfaces deposited purely with C4b (in absence of C3) showed a similar but less pronounced behavior (Figure 4E-F).
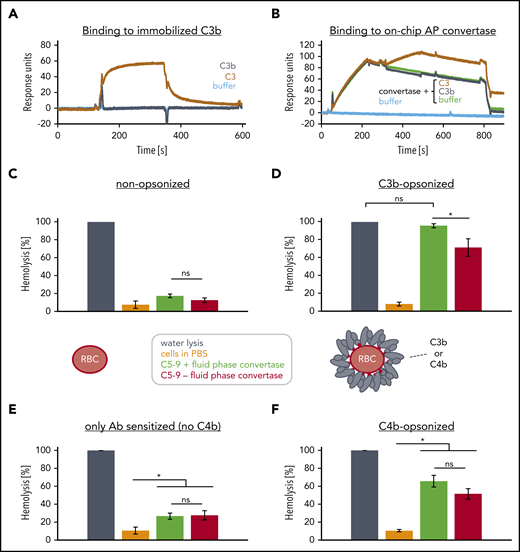
Convertase composition and fluid-phase convertase induced activation of the terminal pathway. (A) Binding of C3 or C3b onto immobilized C3b. A total of 2600 RU of biotinylated C3b (at thioester moiety) was immobilized onto a streptavidin SAP sensor chip and C3 (0.6 µM) or C3b (0.6 µM) were probed for binding. (One typical assay of 2 independent assays is shown.) (B) Binding of C3 or C3b to on chip assembled convertases. 4600 RUs of C3b were immobilized onto a CMD sensor chip. Then a mixture of factor B and factor D were injected (from 20 to 200 seconds) to facilitate the formation of C3bBb convertases before evaluating the binding (at 260-440 seconds) of C3b or C3 to C3bBb. At 740 seconds, the convertases were decayed by injection the decay accelerator CR1(1-3) at 1 µM. (One typical assay of 2 independent assays is shown.) (C-D) Effects of soluble complement components and fluid phase C3bBb convertases on nonopsonized and C3b opsonized rRBC. (C) Washed and nonopsonized and (D) highly dense C3b-opsonized rRBC were mixed with physiological concentrations of a C5-C9 mix and either PBS (red) or a separately prepared fluid-phase convertase C3bBb stored in EDTA buffer (green). The separately prepared soluble C3bBb was assembled by incubating soluble C3b with factor B and factor D in PBS supplemented with 5 mM MgCl2 for 2 minutes before stopping de novo convertase formation by supplementing EDTA to 10 mM. Incubation of cells in water (dark gray) served as reference and was set to 100% lysis; cells incubated in PBS are shown in orange. Released hemoglobin was measured as a marker of hemolysis. The average of 3 independent assays with SD is shown. An ordinary 1-way ANOVA using Tukey's multiple comparisons test was applied. (E-F) Effects of soluble complement components and fluid phase C3bBb convertases on antibody sensitized shRBC that either were or were not opsonized with C4b; otherwise as in panels C and D. ns, not significant.
Convertase composition and fluid-phase convertase induced activation of the terminal pathway. (A) Binding of C3 or C3b onto immobilized C3b. A total of 2600 RU of biotinylated C3b (at thioester moiety) was immobilized onto a streptavidin SAP sensor chip and C3 (0.6 µM) or C3b (0.6 µM) were probed for binding. (One typical assay of 2 independent assays is shown.) (B) Binding of C3 or C3b to on chip assembled convertases. 4600 RUs of C3b were immobilized onto a CMD sensor chip. Then a mixture of factor B and factor D were injected (from 20 to 200 seconds) to facilitate the formation of C3bBb convertases before evaluating the binding (at 260-440 seconds) of C3b or C3 to C3bBb. At 740 seconds, the convertases were decayed by injection the decay accelerator CR1(1-3) at 1 µM. (One typical assay of 2 independent assays is shown.) (C-D) Effects of soluble complement components and fluid phase C3bBb convertases on nonopsonized and C3b opsonized rRBC. (C) Washed and nonopsonized and (D) highly dense C3b-opsonized rRBC were mixed with physiological concentrations of a C5-C9 mix and either PBS (red) or a separately prepared fluid-phase convertase C3bBb stored in EDTA buffer (green). The separately prepared soluble C3bBb was assembled by incubating soluble C3b with factor B and factor D in PBS supplemented with 5 mM MgCl2 for 2 minutes before stopping de novo convertase formation by supplementing EDTA to 10 mM. Incubation of cells in water (dark gray) served as reference and was set to 100% lysis; cells incubated in PBS are shown in orange. Released hemoglobin was measured as a marker of hemolysis. The average of 3 independent assays with SD is shown. An ordinary 1-way ANOVA using Tukey's multiple comparisons test was applied. (E-F) Effects of soluble complement components and fluid phase C3bBb convertases on antibody sensitized shRBC that either were or were not opsonized with C4b; otherwise as in panels C and D. ns, not significant.
Conformational activation of C5 leads to lytic pore formation
To prove that binding of C5 to densely C3b opsonized surfaces that are void of convertases results in conformational C5 activation, we exposed non- and C3b-opsonized rRBCs to C5-7. If C5 can be activated without proteolytic cleavage, the anaphylatoxin domain C5a should be detectable in addition to activated C5 being capable of forming stable C5-7 complexes that insert into the cell membrane without lysing them.37-40 Hence, we stained the treated cells with anti-C3c (as a measure of C3b), an anti-C5a antibody, or fluorescently labeled eculizumab (Figure 5A). Only cells with the highest C3b density were positive for the C5a epitope. Remarkably, fluorescently labeled eculizumab also bound to C5 or C5-7 complexes on the C3b opsonized erythrocytes. To ascertain whether the anti-C5a or eculizumab reactivity on the cells was indeed directed against functional C5-7 complexes, we exposed the cells additionally to C8-9, which resulted in lysis. The remaining cells were analyzed by flow cytometry: only the cells exhibiting the highest C3b density and positive for the C5a epitope underwent lysis (Figure 5B). Hence, C5 can activate without the need for proteolytic processing when bound to highly dense C3b surfaces. To unequivocally prove that conformational C5 activation and C5-9 complex formation can occur purely upon C3b opsonin binding in complete absence of any enzymes, we performed an analysis with purified components on an SPR chip (Figure 5C). Neither a C6-9 mixture, a neoepitope antibody against activated C9, nor a control antibody bound to immobilized C3b. C5 (at 0.375 µM) delivered a prominent binding response with a mix of C5-9 (0.375 µM each) returning a twofold higher binding signal than for C5 alone, suggesting that components from C6-9 have bound to the C3b surface in addition to C5. Finally, the distinct binding response achieved for the anti-C9 neoepitope antibody being probed after the C5-9 mix proved that C9 was captured on the chip surface in its activated conformation, although this SPR surface has never been exposed to a convertase or another enzyme. Another confirmatory experiment demonstrates that freeze-thaw cycles of a mix of C5 and C6 induces conformational activation of C5. As previously reported, such activated C5, denominated C5conf from here on, can form lytic complexes together with C6-9. In such C5conf-9 complexes, the neoepitope present in C5b-9 and the C5a epitope can be substantiated, whereas C5b-9 is only positive for the former epitope (supplemental Figure 6).
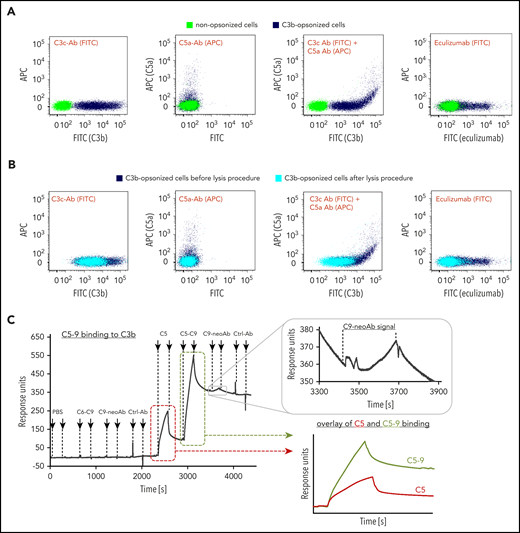
Conformational activation of C5. (A) Presence of C5 on highly dense C3b opsonized rRBCs. Nonopsonized cells (control, green) and C3b opsonized cells (dark blue) were mixed with C5-C7 and stained with a fluorescein isothiocyanate (FITC)-conjugated anti-human C3c mAb as a measure for C3b opsonization (first panel), a biotinylated anti-human C5a Ab (second panel) or simultaneously with both antibodies (third panel). C5 detection was achieved by fluorescence-labeled eculizumab as well (fourth panel). Fluorescence signals were measured by flow cytometry. (One typical assay of 3 independent assays is shown.) (B) Only rRBCs with the highest C3b deposition lysed after allowing lysis. C3b-opsonized cells from panel A carrying C5-7 complexes were exposed to C8 and C9 to enable whole assembly of MAC. After allowing lysis, the remaining cells were stained again with the antibodies (cyan). For better comparison, C3b-opsonized cells (dark blue) from before lysis are shown again. (One typical assay of 3 independent assays is shown.) (C) C5-9 binding to C3b-coated SPR sensor chip. A total of 2000 RUs of biotinylated C3b was immobilized on an SAP sensor chip followed by consecutive injections of buffer, C6-C9, C9-neoAb (anti-C5b-9 mAb), control Ab (anti-6x-His-tag mAb), C5, C5-C9, C9-neoAb, and control Ab. All complement proteins had a concentration of 0.375 µM, the antibodies were injected at 0.05 mg/mL. An overlay of the C5 binding curve (red) and the C5-C9 binding curve (green) was done to point out differences between binding signals. (One typical assay of 2 independent assays is shown.)
Conformational activation of C5. (A) Presence of C5 on highly dense C3b opsonized rRBCs. Nonopsonized cells (control, green) and C3b opsonized cells (dark blue) were mixed with C5-C7 and stained with a fluorescein isothiocyanate (FITC)-conjugated anti-human C3c mAb as a measure for C3b opsonization (first panel), a biotinylated anti-human C5a Ab (second panel) or simultaneously with both antibodies (third panel). C5 detection was achieved by fluorescence-labeled eculizumab as well (fourth panel). Fluorescence signals were measured by flow cytometry. (One typical assay of 3 independent assays is shown.) (B) Only rRBCs with the highest C3b deposition lysed after allowing lysis. C3b-opsonized cells from panel A carrying C5-7 complexes were exposed to C8 and C9 to enable whole assembly of MAC. After allowing lysis, the remaining cells were stained again with the antibodies (cyan). For better comparison, C3b-opsonized cells (dark blue) from before lysis are shown again. (One typical assay of 3 independent assays is shown.) (C) C5-9 binding to C3b-coated SPR sensor chip. A total of 2000 RUs of biotinylated C3b was immobilized on an SAP sensor chip followed by consecutive injections of buffer, C6-C9, C9-neoAb (anti-C5b-9 mAb), control Ab (anti-6x-His-tag mAb), C5, C5-C9, C9-neoAb, and control Ab. All complement proteins had a concentration of 0.375 µM, the antibodies were injected at 0.05 mg/mL. An overlay of the C5 binding curve (red) and the C5-C9 binding curve (green) was done to point out differences between binding signals. (One typical assay of 2 independent assays is shown.)
Nonenzymatic C5 activation in presence of C5 inhibitors
Neither Coversin nor eculizumab can completely inhibit C5 and thus TP activity during conditions of strong complement activation.15,17 Eculizumab bound to nonenzymatically activated C5 molecules entrapped in C5-7 complexes (Figure 5 A-B), which is in line with observations in a clinical diagnostic setting.10 The more important question remains whether eculizumab binding to C5 is sufficient to block nonenzymatic conformational C5 activation. Coversin and eculizumab address different binding patches on opposite sites on C5 (Figure 6A-B).31,41 Although Coversin binds 3 C5 domains, which are close in space in nonactivated C5 but far apart in C5b (supplemental Figure 7A), eculizumab binds to the single MG-7 domain in C5, which changes its relative conformation during transition from C5 to C5b but keeps its overall structure conserved (supplemental Figure 7B). Our SPR assay shows that Coversin exclusively binds to native (nonactivated) C5, whereas eculizumab binds with virtually identical kinetics to both activated and native C5, corroborating that C5b complexes may compete with C5 for eculizumab binding (Figure 6C-D). Evaluating the inhibition of conformational activation showed that Coversin and eculizumab both reduced nonenzymatic C5 activation to similar extents, but substantial levels of residual hemolysis remained (Figure 6E). Only simultaneous double C5 inhibition by Coversin and eculizumab reduced conformational C5 activation to baseline levels. Previous reports studying C5 inhibition demonstrated that eculizumab reached higher efficiency in inhibiting C5 activation compared with Coversin.15,17 Hemolysis assays in NHS under C5 blockage are envisaged to result in both canonical convertase-mediated and conformational C5 activation (because under C5 block, high densities of C3b can be achieved). Under these conditions, eculizumab may have a benefit over Coversin. Although both C5 inhibitors stabilize an unproductive C5 conformation, eculizumab binds close to the convertase cleavage site and appears to additionally block convertase access through steric hindrance.31,41
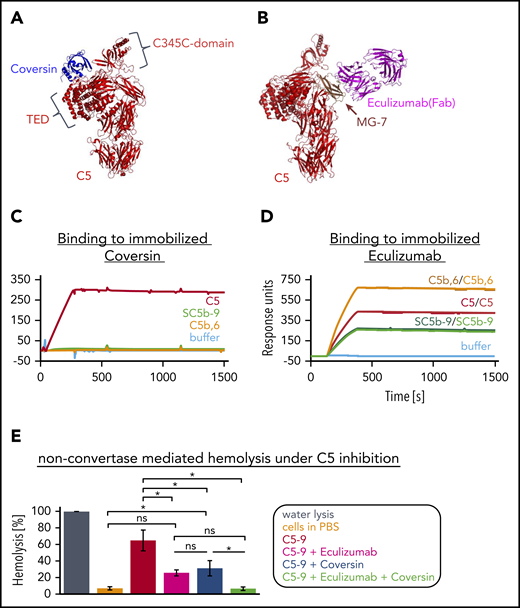
Conformational activation of C5 in presence of C5 inhibitors. (A) Structure of the complex of C5 and Coversin. Crystal structure of C5:Coversin (PDB entry: 5HCC Jore et al31 ). Coversin addresses 3 domains in C5: CUB, TED, and the C345C domain (with the latter 2 being indicated). (B) Binding of eculizumab to C5. Crystal structure of eculizumab-Fab (pink) in complex with C5 (red) (PDB entry: 5I5K Schatz-Jakobsen et al41 ). Eculizumab binds solely to the MG-7 domain (brown) of C5. (C) Binding curves of C5 and activated C5 complexes to 440 RU of immobilized Coversin (via amine coupling onto a CMD sensor chip). C5 (red) and the activated C5 structures, C5b,6 (orange) and SC5b-9 (green), were applied to the chip at a concentration of 25 nM. (One assay was performed.) (D) Binding curves of C5 and activated C5 complexes to eculizumab immobilized via amine coupling to a level of 13,200 RUs onto a CMD sensor chip. All analytes were injected at a concentration of 25 nM. Duplicates of each sample were performed to show reproducibility. (One assay was performed.) (E) Nonconvertase-mediated hemolysis under C5 inhibition. C3b-opsonized rRBCs were exposed to a C6-C9 mixture (physiological concentrations), followed by the addition of C5 (physiological concentration) preincubated with eculizumab (5 µM, pink), Coversin (5 µM, blue), both C5 inhibitors together (green) or without inhibitor (red). Cells in water served as reference and were set to 100% lysis, incubation of cells only in PBS is shown in orange. Release of hemoglobin was measured as a marker of hemolysis. Average of 3 independent assays with SD is shown. An ordinary 1-way ANOVA using Tukey multiple comparisons test was applied. ns, not significant.
Conformational activation of C5 in presence of C5 inhibitors. (A) Structure of the complex of C5 and Coversin. Crystal structure of C5:Coversin (PDB entry: 5HCC Jore et al31 ). Coversin addresses 3 domains in C5: CUB, TED, and the C345C domain (with the latter 2 being indicated). (B) Binding of eculizumab to C5. Crystal structure of eculizumab-Fab (pink) in complex with C5 (red) (PDB entry: 5I5K Schatz-Jakobsen et al41 ). Eculizumab binds solely to the MG-7 domain (brown) of C5. (C) Binding curves of C5 and activated C5 complexes to 440 RU of immobilized Coversin (via amine coupling onto a CMD sensor chip). C5 (red) and the activated C5 structures, C5b,6 (orange) and SC5b-9 (green), were applied to the chip at a concentration of 25 nM. (One assay was performed.) (D) Binding curves of C5 and activated C5 complexes to eculizumab immobilized via amine coupling to a level of 13,200 RUs onto a CMD sensor chip. All analytes were injected at a concentration of 25 nM. Duplicates of each sample were performed to show reproducibility. (One assay was performed.) (E) Nonconvertase-mediated hemolysis under C5 inhibition. C3b-opsonized rRBCs were exposed to a C6-C9 mixture (physiological concentrations), followed by the addition of C5 (physiological concentration) preincubated with eculizumab (5 µM, pink), Coversin (5 µM, blue), both C5 inhibitors together (green) or without inhibitor (red). Cells in water served as reference and were set to 100% lysis, incubation of cells only in PBS is shown in orange. Release of hemoglobin was measured as a marker of hemolysis. Average of 3 independent assays with SD is shown. An ordinary 1-way ANOVA using Tukey multiple comparisons test was applied. ns, not significant.
Discussion
Stoichiometric inhibitors of the central complement components C3 and C5 are building a hotspot in current complement intervention strategies.9 Here, we demonstrate that the complement cascade proceeds differently than assumed with important implication for therapeutic intervention strategies at the levels of C3 and C5.
We show that triggering the CP in complete absence of C3 or when C3 is inhibited still leads to considerable C5 activation and lytic TP activity (Figure 1; supplemental Figure 1). We establish that C3-bypass activation is the main reason for these observations, with a second reason being the differential inhibition of C3 activation by Compstatin after CP activation, which in contrast to AP-mediated C3 activation, is not 100% complete (Figures 1E-F and 2E-F). Establishing the underlying mechanism of the C3-bypass activation of C5 underscores that this activation route is a general phenomenon. We confirm that additional C3b molecules produced by the bimolecular convertase C3bBb recruit and prime C5 for cleavage by the bimolecular C3bBb convertase similarly as proposed before.29 In addition, we demonstrate that additionally produced C3b does not associate with C3bBb, challenging the dogma that C5 convertases comprise a trimolecular complex (Figure 4A-B).
Instead, deposited and thus clustered C3b molecules have 2 binding sites for C5: the MG4-MG5 site as shown before33,34 and a site on C3b that gets lost upon binding of Bb or degradation of C3b into iC3b (Figures 2B and 3C-D; supplemental Figures 4 and 8A). The C3b homolog C4b shares important functions with C3b: C3b and C4b both bind C3 and C5 with estimated low micromolar affinity (Figure 2A-D). Hence, it is not surprising that either purely C3b decorated (or C4b decorated) cells can recruit and prime C5 for cleavage by the fluid phase bimolecular convertase C3bBb (Figure 4C-F). Very high densities of C3b or C4b, however, can directly activate C5 without proteolytic cleavage of the anaphylatoxin domain C5a, which remains detectable within a functional MAC pore leading to the formation of C5conf-9 in absence of convertases (Figure 5A-C; supplemental Figure 6). It is well documented that chemical (low pH,42,43 oxygen radicals44,45 ) or physical stress (freeze-thawing21 ) induces nonenzymatic C5 activation, yielding C5b-like properties and in consequence reactive lysis (similarly as C5b-6 complexes39,46 do). We propose that prolonged interactions with a densely deposited C3b and/or C4b also induces a conformational change in C5 comparable to C3 transitioning to C3(H2O).47,48 In this way activated C5 may be termed C5conf because conformational activated C5 displays C5b-like properties but still contains C5a (supplemental Figure 8B).
Importantly, these new insights into C5 activation provide the rational why (1) different stoichiometric C5 inhibitors fail to completely inhibit lytic TP activity, (2) why the level of TP inhibition by different inhibitors can vary, and (3) why C5 double inhibition reliably prevents C5 activation. Generally, upon C5 inhibitor binding, the native C5 conformation becomes stabilized, hindering C5 priming for subsequent convertase cleavage but inhibition of C5 priming correlates negatively with the surface density of C3b or C4b. Indeed, very high C3b/C4b surface densities alone can induce conformational activation of C5 to C5conf, a process that can be hindered but not completely blocked by single stoichiometric C5 inhibitors. In addition to blocking of C5 priming, inhibitors like eculizumab that bind to the vicinity of the convertase cleavage site also block convertase access to primed C5; hence, eculizumab is a more active inhibitor than Coversin in vitro17,22,36 and in the clinical setting.16 Ideally, 2 inhibitory mechanisms are unified in 1 stoichiometric C5 inhibitor: blocking the conformational priming/activation of C5 and the steric access to the convertase unit (or as another possibility, blocking the assembly of MAC complexes as seen for RO711268949 ). However, the only way to totally inhibit C5 activation appears to be when 2 stoichiometric C5 inhibitors are used simultaneously and thus stably fix C5 in its unproductive, nonprimed conformation (Figure 6E). Remarkably, we uncovered that eculizumab binds almost identically to native C5- and C5b-containing complexes, indicating a potential competition with C5. Moreover, these data explain why eculizumab deposits in vessel walls in thrombotic microangiopathy with a similar distribution as C5b-9.50
Our findings have major clinical implications. The uncovered C3-bypass activation mechanism of C5 challenges the conclusions from numerous in vivo studies using C3-deficient animals to interrogate whether complement activation is the driver of disease pathology in the analyzed models. In all these disease models, it was mechanistically assumed (according to dogma) that C3 knockout abolishes practically all complement effector functions. When C5 activation products have been detected despite using C3-knockout animals, this was often attributed to extrinsic pathway activation.51,52 However, contrary to dogma, strong CP (and/or LP) activation, which leads to dense C4b deposition, can still promote C5 activation with all its cell-damaging consequences.24 Hence, disease models (eg, for autoimmune diseases) with established, strong involvement of the CP/LP that failed to produce a benefit upon C3 depletion (eg, with Cobra-venom-factor or by C3 gene deletion) cannot rule out that disease pathology is driven via intrinsic CP/LP activation leading to TP effector functions; thus, such studies may have to be reevaluated. Auger et al identified that autoantibody-mediated arthritis is still driven by C5 activation despite the absence of C3 in the animals.53 Shapiro et al observed that antifungal antibodies protected mice in C3 knockout mice, although it is known that complement activation is important for defense against Cryptococcus neoformans.54 Finally, in view of the C3-bypass activation route of C5, the use of stoichiometric C3 inhibitors in CP-driven diseases may need to be reconsidered. Of note, several published reports in human ex vivo or animal models demonstrate residual terminal pathway activity when C3 is blocked by a peptide inhibitor.55-57 Residual TP activity likely inflicts considerably less damage to nucleated (eg, endothelial) cells relative to RBCs, which are especially susceptible to MAC. But even sublytic/sublethal MAC on nucleated cells was shown to lead to cell-activation effects like cell swelling, energy expenditure, and inflammatory signaling (supplemental Figure 1D-F).58,59 The finding on residual C5 activity in absence of C3 also affects the safety aspects of stoichiometric C3 inhibitors (eg, although Compstatin either nearly completely or totally blocks CP- or AP-mediated C3 activation and thus C3b opsonization, respectively, C4b deposition and TP effector functions after strong CP/LP activation remain possible). These findings signify that C3 peptide inhibitors may feature a broader safety profile than previously thought. Apart from the clinical implications, these findings also propose that the complement pathways appear to function slightly differently than thought. Figure 7 and supplemental Figure 9 illustrate complement schemes consistent with the findings of this study.
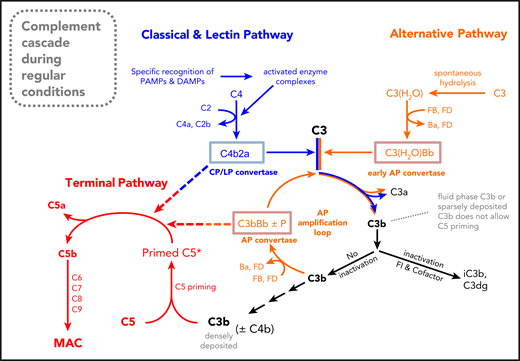
A new schematic drawing of the complement cascade. The new feature of this complement scheme is the absence of trimolecular C5 convertases. Instead, it is envisaged that the bimolecular C3 convertases C3bBb (or C3bBbP) and C4b2a proteolytically activate C5 once it is conformationally prepared for cleavage (or primed) on highly dense surface clusters of C3b alone or C3b and C4b.
A new schematic drawing of the complement cascade. The new feature of this complement scheme is the absence of trimolecular C5 convertases. Instead, it is envisaged that the bimolecular C3 convertases C3bBb (or C3bBbP) and C4b2a proteolytically activate C5 once it is conformationally prepared for cleavage (or primed) on highly dense surface clusters of C3b alone or C3b and C4b.
Regarding C5 activation and inhibition, we show that C5 activation can proceed without proteolytic activation by a convertase on surfaces that are densely deposited with C3b or C4b. Paradoxically, high C3b/C4b opsonin densities only occur during C5 inhibition and concomitant complement activation. Without C5 blockage, cells would be quickly destroyed/damaged via TP-mediated lysis before they fix even moderate amounts of C3/C4b opsonins. Because residual TP activity is based on the ability of C5 to activate purely through conformational change upon interacting with its opsonin ligands, it must be assumed that residual C5 activity under C5 inhibition is a class effect of most stoichiometric C5 inhibitors.
For original data, please e-mail the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Anke Schultze for her excellent technical assistance.
This work was supported by Deutsche Forschungsgemeinschaft grants SCHM 3018/2-1 (C.Q.S.) and INST 40/479/2, CRC1149 (M.H.-L.).
Authorship
Contribution: C.Q.S. and H.S. conceived the study; M.M., A.D., S.J.L., R.H., and C.Q.S. planned and performed the experiments; M.M., A.D., C.Q.S., O.Z., C.K.B., and H.S. analyzed the data; A.S., M.H.-L., B.H., O.Z., C.K.B., and H.S. advised on experimental design and data analysis; M.M., A.D., A.S., B.H., C.K.B., and C.Q.S. prepared important reagents; all authors contributed to the discussion of the data; C.Q.S., M.M., M.H.-L., and A.D. wrote the manuscript; and all authors critically revised the manuscript.
Conflict-of-interest disclosure: C.Q.S. and M.H.-L. are inventors of patent applications that describe the use of complement inhibitors for therapeutic applications. C.Q.S. has received research funding from Takeda Pharmaceutical. M.H.-L., B.H., C.Q.S., and H.S. received honoraria for speaking at symposia organized by Alexion Pharmaceuticals. H.S. and B.H. served on an advisory committee for and received research funding from Alexion Pharmaceuticals. H.S. served on an advisory committee for Ra Pharmaceuticals and Alnylam. The remaining authors declare no competing financial interests.
The current affiliation for O.Z. is Brandenburg Medical School Theodore Fontane, Institute of Clinical Pharmacology, Immanuel Klinik Rüdersdorf, Rüdersdorf, Germany.
Correspondence: Christoph Q. Schmidt, The Institute of Pharmacology of Natural Products and Clinical Pharmacology, Ulm University, Helmholtzstraße 20, D-89081 Ulm, Germany; e-mail: christoph.schmidt@uni-ulm.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal