

In this issue of Blood, have identified a “C3 bypass” activation of the complement component C5.1 On highly opsonized surfaces, the cell-killing membrane attack complex (MAC) may be formed without a C5 convertase, because of a conformational change in C5, which adopts a C5b-like structure. These findings may explain why C5 inhibitors do not always prevent residual C5 activity in patients with massive complement activation.
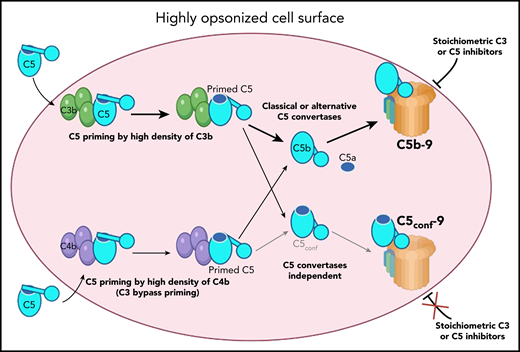
Proposed model for MAC formation on surfaces, opsonized with C3b or C4b (such as pathologic red blood cells): C5, coming from the circulation (cyan), binds to one of the many C3b (green) or C4b (violet) molecules on the cell surface (rose). The interaction induces conformational change by moving the thioester domain (blue circle in C5) partially down, and exposure of the C5a domain (blue ellipse), to facilitate its cleavage by the C5 convertases. After such proteolytic activation, C5a is released, and the thioester domain in C5b goes down to its maximum extent, exposing the C6 binding site and allowing the formation of the C5b-9 MAC. This process will be blocked by stoichiometric C3 or C5 inhibitors, explaining their therapeutic effect. In the context of very high densities of C3b or C4b, the interaction with C5 is stronger, allowing full transition of the thioester domain down, assuming a C5b-like structure, without cleavage and release of the C5a domain. This newly formed C5conf will have the C6 binding site exposed and the capacity to form a C5conf-C9 MAC. This new process is expected to occur rarely, only at very high density of C3b or C4b and in the absence of active convertases. The formation of this C5conf-9 membrane attach complex, though, will not be prevented by stoichiometric C3 or C5 inhibitors and may contribute to the pharmacodynamic breakthrough effect. Illustration by Margot Revel, Centre de Recherche des Cordeliers, using BioRender.com.
Proposed model for MAC formation on surfaces, opsonized with C3b or C4b (such as pathologic red blood cells): C5, coming from the circulation (cyan), binds to one of the many C3b (green) or C4b (violet) molecules on the cell surface (rose). The interaction induces conformational change by moving the thioester domain (blue circle in C5) partially down, and exposure of the C5a domain (blue ellipse), to facilitate its cleavage by the C5 convertases. After such proteolytic activation, C5a is released, and the thioester domain in C5b goes down to its maximum extent, exposing the C6 binding site and allowing the formation of the C5b-9 MAC. This process will be blocked by stoichiometric C3 or C5 inhibitors, explaining their therapeutic effect. In the context of very high densities of C3b or C4b, the interaction with C5 is stronger, allowing full transition of the thioester domain down, assuming a C5b-like structure, without cleavage and release of the C5a domain. This newly formed C5conf will have the C6 binding site exposed and the capacity to form a C5conf-C9 MAC. This new process is expected to occur rarely, only at very high density of C3b or C4b and in the absence of active convertases. The formation of this C5conf-9 membrane attach complex, though, will not be prevented by stoichiometric C3 or C5 inhibitors and may contribute to the pharmacodynamic breakthrough effect. Illustration by Margot Revel, Centre de Recherche des Cordeliers, using BioRender.com.
“There are only two little clouds in the sky of physics,” said Lord Kelvin, referring to the completeness of knowledge in classical physics at start of the 20th century. With this witticism, he pointed to the directions needing additional work. Indeed, solving the problems behind the “two little clouds” gave rise to the theory of relativity and quantum mechanics: 2 pillars of modern physics. If there are “two little clouds in the sky” of the complement system today, they are related to C5 convertase and to intracellular complement.
Complement is an innate immune defense system, protecting us against pathogen infections and contributing to the host homeostasis.2 Nevertheless, when inappropriately overactivated, it causes cell damage, resulting in severe and life-threatening diseases. The effectors of the terminal complement pathway are C5a, a very potent proinflammatory mediator, and the MAC C5b-9, which creates pores in the cell membrane, lysing bacteria, and red blood cells or activating nucleated host cell.2 It is traditionally accepted that they are generated after cleavage of C5 by an enzymatic complex called C5 convertase. Early studies suggested that C3 convertase of the classical or alternative complement pathways becomes C5 convertase after the covalent binding of C3b directly to the preexisting C3 convertase or to the cell surface in the immediate proximity of a C3 convertase.3,4 This was thought to induce a change in the specificity of the enzymes, stopping cleavage of C3 and starting cleavage of C5. The current understanding is that high density of C3b will result in a transient C3b-C5 interaction, leading to a conformational change in C5 (C5 priming), making it capable of binding with C3 convertases.5,6 The study of Mannes et al refines this model, showing that at high density, C4b can also prime C5 (see figure). This observation may challenge studies, where the contribution of complement was ruled out if C3-deficient mice were not protected in the particular disease model being tested. The authors argue that even in absence of C3, high density of C4b on target surfaces could trigger the terminal pathway and still induce cell damage in a C3 bypass pathway.
Strikingly, the authors reveal that at very high surface density of C3b or C4b, MAC could form in the absence of C5 activating enzymes, that is, in a C5 convertases bypass pathway.1 They explain this finding by a conformational change, in which C5 adopts a C5b-like structure (see figure). This structural modification resembles the one of C3, which changes conformation upon hydrolysis of its thioester bond, becoming C3(H2O). This C5b-like C5 (named C5conf) will interact with C6, C7, C8, and C9 to form an MAC.
Recent discoveries about the functioning of the C5 convertases have often been made because of studies on the mechanism of action of therapeutic C5 inhibitors.1,6 C5 blockade has revolutionized the treatment of many diseases of complement overactivation, including, but not limited to, paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome.7 Experience with the use of the pioneering C5 inhibitor eculizumab revealed the clinical phenomenon of residual C5 activity, even with an excess of the C5 blocker (pharmacodynamic breakthrough). With the advent of novel complement inhibitors, acting at every stage of the cascade, it is of crucial importance to understand the mechanisms of the activity of this residual terminal pathway and how it will be impacted by these novel drugs. Mannes et al show that not only C5 but also stoichiometric C3 inhibition alone is not able to completely protect from terminal pathway activity and cell lysis and that combination approaches are needed for a successful terminal pathway control (see figure).
These discoveries raise many questions of pathophysiological relevance. In which condition does this occur in vivo? Could it impact nucleated host cells? Does it contribute to bacterial elimination? Is it possible to counteract it with complement inhibitors on host cells or accelerate it at the bacterial surface? If C5conf can form MAC, it could potentially explain the presence of persistent “sC5b-9” in some patients treated with eculizumab. At least in part, this sC5b-9 could be in fact sC5conf-9. This could be tested by detection of a C5a reactivity (presence of the uncleaved ANA domain) in such complexes. Interestingly, conformation change of C5 (presumably the same as C5conf) was previously shown to expose a C5a neoepitope without C5a fragment formation.8 Such conformational change could occur also after binding of eculizumab.8 To what extent could this eculizumab-C5 form C5conf-9? Why do eculizumab deposits cooccur with the ones of MAC in the vessel walls in thrombotic microangiopathy patients9 ? What is the exact structure of C5 convertases and how are they different, especially when compared with the C3 convertases? Does intracellular C5, proposed as the controller of inflammasome assembly in T cells,10 need to adopt a primed conformation in order to exert its functions? Could a single C5 inhibitor achieve fixation of its target protein in an unproductive, nonprimed conformation? How, with a combination of the currently available complement inhibitors, could one prevent the pharmacodynamic breakthrough and its deleterious effects in patients? All these and many other questions are a direction for future work before clearing the C5 convertase cloud from the sky of the complement biology.
In summary, the study of Mannes et al provides a novel fundamental basis for explanation of the terminal complement pathway activation, where C5 priming by C3b or C4b is a prerequisite for enzymatic and nonenzymatic C5 activation and that this conformational activation of C5 leads to lytic pore formation. These findings may explain, at least in part, the pharmacodynamic breakthrough of complement inhibitors in paroxysmal nocturnal hemoglobinuria or other diseases, where densely opsonized cells (especially red blood cells) accumulate. These findings open new perspectives for development and optimization of the use of complement inhibitors in the clinical practice.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal