

Key Points
Emicizumab has good hemostatic efficacy in AHA: within a few days after the first injection, less bypassing therapy is needed.
Low emicizumab concentrations can prevent breakthrough bleeding: outpatient patient management with visits every 1 to 3 weeks is feasible.

Abstract
Acquired hemophilia A (AHA) is a severe bleeding disorder caused by inhibiting autoantibodies to coagulation factor VIII (FVIII). For hemostatic treatment, bypassing agents and human or porcine FVIII are currently standard of care. Emicizumab is a bispecific, FVIII-mimetic therapeutic antibody that reduced the annualized bleeding rates in congenital hemophiliacs. Here, we report on 6 male and 6 female patients with AHA treated with emicizumab (all data medians and interquartile range), age 74 (64-80) years, initial FVIII <1%; inhibitor titer 22.3 Bethesda units (BU)/mL (range, 3-2000). Eight patients had severe bleeding. Emicizumab was started, 3 mg/kg subcutaneously, weekly for 2 to 3 doses, followed by 1.5 mg/kg every 3 weeks to keep the lowest effective FVIII levels. For FVIII monitoring, chromogenic assays with human and bovine reagents were used. All patients received immunosuppression with steroids and/or rituximab. After the first dose of emicizumab, activated partial thromboplastin time normalized in 1 to 3 days, FVIII (human reagents) exceeded 10% after 11 (7.5-12) days. Hemostatic efficacy was obtained and bypassing therapy stopped after 1.5 (1-4) days. FVIII (bovine reagents) exceeded 50%, indicating complete remission after 115 (67-185) days, and emicizumab was stopped after 31 (15-79) days. A median of 5 injections (range, 3-9) were given. No patient died of bleeding or thromboembolism, and no breakthrough bleeding was observed after the first dose of emicizumab. In conclusion, emicizumab seems to be an effective hemostatic therapy for AHA, with the advantages of subcutaneous therapy, good hemostatic efficacy, early discharge, and reduction of immunosuppression and adverse events.
Introduction
Acquired hemophilia A (AHA) is a severe bleeding disorder caused by autoantibodies inhibiting the function of coagulation factor VIII (FVIII).1-5 It is characterized by the new onset of bleeding in 90% of affected patients, which is severe in 70% of them.6,7 AHA mainly affects patients of higher age: the median age reported in the European Acquired Haemophilia Registry-2 (EACH2) was 74 years.6 Most of the patients have considerable comorbidities. For initial hemostatic treatment, bypassing agents (recombinant activated human FVII [rhFVIIa], activated prothrombin complex concentrates [APCC]) are standard of care and are highly effective in treating the bleeding.7-10 In the EACH2 registry, efficacy rates of 93% are reported.7 FVIII replacement with human FVIII concentrates, either plasma-derived or recombinant, is effective only with low inhibitor titers, with efficacy rates of ∼70%.7,9,10 The use of recombinant porcine-sequence FVIII concentrates is an option for patients without cross-reactivity.11,12 All of these therapies, although effective, have the disadvantages of frequent IV infusions, high costs, and some risk of thromboembolic complications. The rate of fatal bleeding has been reduced to 3% by the current hemostatic therapies; the rate of thromboembolic complications is in the range of 0% to 4.8%.6,7,10,13,14
The causal treatment of AHA, however, is to stop the underlying autoimmune process and eradicate the autoantibodies. Immunosuppression with corticosteroids with or without cytotoxics like cyclophosphamide have been used for many years, but is associated with a high rate of severe, even fatal, adverse effects.15,16 Especially in the elderly population, highly vulnerable comorbid patients are prone to severe complications. A trial of the Gesellschaft für Thrombose und Hämostaseforschung (GTH; GTH-AHA 01/2010 trial) demonstrated that even an escalating immunosuppressive approach is not able to obtain faster remissions and save the costs of hemostatic therapy, but rather causes harm to the patients16,17 : up to 16% of patients in that trial died of infections or other immunosuppression-related events. The addition of rituximab may reduce such unacceptable adverse effects, but is not licensed for this indication and time to remission may be longer.2,3,18-21 Factors predicting the outcome of AHA have been identified in the GTH-AH 01/2010 trial (low initial FVIII activity, high inhibitor titers, immunoglobulin A [IgA] autoantibodies to FVIII, poor World Health Organization [WHO] performance state, coexisting malignancy),16,17,22 which opened the possibility of estimating the course of the disease, the time at risk of bleeding, and the expected duration of immunosuppression.
Emicizumab is a bispecific, FVIII-mimetic therapeutic antibody that has considerably reduced the annualized bleeding rates in congenital hemophiliacs with and without inhibitors with weekly or even 3- to 4-weekly subcutaneous treatment.23-27 Emicizumab is already approved for prophylaxis of bleeding in hemophiliacs with and without inhibitors, and it is commercially available. Preclinical data and the pathophysiological concept also suggest advantages in AHA. Single-case reports on the use of emicizumab in patients with AHA have been published in the last year.28-30 However, there are several points that need special attention when treating such patients with emicizumab: interference of laboratory testing with concurrent patient’s own FVIII in the presence of inhibiting autoantibodies and/or bypassing agents, potential thrombogenicity, detection of remission, and necessary dose and intervals are almost unknown.
Here, we present a series of 12 patients with AHA, in whom emicizumab has been used as hemostatic therapy.
Patients and methods
We report on a series of 12 patients with newly diagnosed AHA, admitted to the Department of Hematology of the University Hospital of Vienna, in whom emicizumab was used as a hemostatic agent. All patients had a prolonged activated partial thromboplastin time (APTT) and had experienced new-onset bleeding that led to the diagnosis of AHA. Table 1 shows the characteristics of these patients. Six were female and 6 male, and median age was 74 years (range, 51-87 years). Severe bleeding (organ-, limb- or life-threatening, drop in hemoglobin levels >2 g/dL, or need for ≥2 red blood cell transfusions) was observed in 8 of the patients; 6 of them had associated major surgery and bleeding from surgical wounds. Deep muscle hematomas were found in 5 patients, superficial muscle hematomas in 10 patients, retroperitoneal bleeding in 1, and all patients had extended skin hematomas (which had occurred spontaneously).
Demography and patient’s characteristics
| Median | IQR | Range | |
|---|---|---|---|
| Age at diagnosis, y | 74 | 65-80 | 51-87 |
| Sex, male/female | 6/6 | ||
| Initial bleeding site,*n | |||
| Skin hematomas | 12 | ||
| Deep muscle hematoma (psoas) | 5 | ||
| Superficial muscle | 10 | ||
| Retroperitoneal | 1 | ||
| Surgical wound | 6 | ||
| Hematuria | 1 | ||
| Mucosal bleeding | 2 | ||
| Joint bleedings | 1 | ||
| Central nervous system | 0 | ||
| Gastrointestinal bleeding | 0 | ||
| Trigger of bleeding,*n | |||
| None (spontaneous) | 11 | ||
| Surgery | 6 | ||
| Severity of bleeding | |||
| Severe | 8 | ||
| Mild | 4 | ||
| Time bleeding to diagnosis, d | 4 | 3-6 | 1-36 |
| Medical history,*n | |||
| None | 0 | ||
| Chronic inflammatory bowel disease | 1 | ||
| Metabolic syndrome, adiposity, diabetes | 6 | ||
| Infection, inflammation | 2 | ||
| Vascular disease (heart, peripheral, stroke) | 6 | ||
| Malignant disease | 3 | ||
| Lung disease | 4 | ||
| Psychiatric disorder | 5 | ||
| Initial FVIII activity, % | <1 | <1-1.5 | <1-11 |
| Maximum inhibitor titer, BU/mL | 22.3 | 9-80 | 3.5-2000 |
| Maximum anti-FVIII IgG, AU/mL | 720 | 393-935 | 138-3108 |
| Median | IQR | Range | |
|---|---|---|---|
| Age at diagnosis, y | 74 | 65-80 | 51-87 |
| Sex, male/female | 6/6 | ||
| Initial bleeding site,*n | |||
| Skin hematomas | 12 | ||
| Deep muscle hematoma (psoas) | 5 | ||
| Superficial muscle | 10 | ||
| Retroperitoneal | 1 | ||
| Surgical wound | 6 | ||
| Hematuria | 1 | ||
| Mucosal bleeding | 2 | ||
| Joint bleedings | 1 | ||
| Central nervous system | 0 | ||
| Gastrointestinal bleeding | 0 | ||
| Trigger of bleeding,*n | |||
| None (spontaneous) | 11 | ||
| Surgery | 6 | ||
| Severity of bleeding | |||
| Severe | 8 | ||
| Mild | 4 | ||
| Time bleeding to diagnosis, d | 4 | 3-6 | 1-36 |
| Medical history,*n | |||
| None | 0 | ||
| Chronic inflammatory bowel disease | 1 | ||
| Metabolic syndrome, adiposity, diabetes | 6 | ||
| Infection, inflammation | 2 | ||
| Vascular disease (heart, peripheral, stroke) | 6 | ||
| Malignant disease | 3 | ||
| Lung disease | 4 | ||
| Psychiatric disorder | 5 | ||
| Initial FVIII activity, % | <1 | <1-1.5 | <1-11 |
| Maximum inhibitor titer, BU/mL | 22.3 | 9-80 | 3.5-2000 |
| Maximum anti-FVIII IgG, AU/mL | 720 | 393-935 | 138-3108 |
n = 12.
AU, arbitrary unit; BU, Bethesda unit; IQR, interquartile range.
Some patients had >1 condition.
All patients had comorbidities that prevented intensive immunosuppressive therapy (Table 1): older age (6 patients were older than 75 years); 1 had extensive chronic inflammatory bowel disease, heavily pretreated with >10 abdominal surgeries and long-term immunosuppression, complicated by pulmonary tuberculosis; 5 patients had metabolic syndrome with adiposity, diabetes, and hypertension; 2 had active infections (peritonitis, tuberculosis, large infected abdominal wound); and 6 had severe arteriosclerosis, coronary heart disease, femoral arterial bypass grafting, and atrial fibrillation. More details of the patients are summarized in supplemental Table 1 (available on the Blood Web site). Initial FVIII activity was <1% in 8 patients and the inhibitor titer was >20 Bethesda units (BU)/mL in 6 patients, suggesting a median time to remission of >50 days with steroids plus or minus cyclophosphamide and a high risk for complications according to the risk factors identified in the GTH-AH 01/2010 study.16 For these medical reasons, an individualized therapeutic approach according to the existing active dedicated standard operating procedures for AHA at our institution was used.
Laboratory methods
FVIII coagulation activity (FVIII:C) was measured in triplicate using 3 different dilutions with a 1-stage clotting assay, using the APTT-FS reagent and an FVIII-deficient plasma from Technoclone (Vienna, Austria); normal range was 60% to 180%.
After start of emicizumab, FVIII activity was measured with the chromogenic Biophen FVIII:C assay from Hyphen (Neuville-sur-Oise, France) using human reagents (FVIII:h), which detect the patient’s own FVIII and are also sensitive to the effects of emicizumab (normal range, 50% to 200%). Results were available on the same day and used for dose modifications. To measure absolute emicizumab plasma concentrations did not seem to be useful in patients with AHA considering the interference of the FVIII activity-based assays with the patient’s endogenous FVIII levels.
In addition, a chromogenic FVIII activity assay using bovine reagents (FVIII:b, FVIII chromogenic; Siemens Healthcare Diagnostic Products, Marburg, Germany) was used to detect the patient’s FVIII, but was insensitive to emicizumab (normal range, 70% to 150%).
Prior to emicizumab dosing, FVIII inhibitor titers were quantified with the Nijmegen-modified Bethesda method using the 1-stage FVIII clotting assay (normal range, <0.4 BU/mL). After start of emicizumab, the chromogenic FVIII activity assay using bovine reagents was used for the Bethesda assay.
All FVIII activity measurements were performed on a Siemens CS-5100 analyzer (Siemens, Marburg, Germany). Anti-FVIII IgG concentration was measured with an enzyme-linked immunosorbent assay (ELISA; Zymutest Anti FVIII, Hyphen BioMed) (normal range, <12 arbitrary units [AU]/mL). APTT was measured with the APTT Automate 5 reagent on a STA-Max2 analyzer (Diagnostica Stago, Asneres, France; normal range, 27-41 seconds). Serum chemistry, blood cell counts, D-dimer, and fibrinogen levels were quantified with local standard laboratory methods.
Hemostatic therapy
Initial hemostatic treatment with bypassing agents was started in all patients with severe bleeding according to current guidelines.5 After thorough assessment of the patient’s cases and risk factors (well known as influencing AHA patient outcome, and including severity of bleeding, FVIII level, inhibitor titer, age, comorbidity, response to first-line hemostatic therapy), a board consisting of hematology and hemostasis experts, well experienced in the treatment of hemophilia and bleeding disorders, discussed the cases. Five patients had insufficient response to bypassing therapy, 1 had an adverse event to bypassing therapy, and 3 had social reasons as additional factors limiting the use of approved hemostatic therapy. Five patients had very high inhibitor titers >60 BU/mL, 4 of them after recent surgery, suggesting a prolonged need for hemostatic therapy and a high economic burden. A conclusion that conventional AHA therapy seemed to be associated with a high rate of complications, and that emicizumab plus reduced-intensity immunosuppression could be a better option, led to a shared decision-making process between physicians and patients. Patients were informed about the characteristics of their disease and the advantages and disadvantages of possible and available treatment options. The new options, emicizumab and rituximab, were also explained in detail, as well as the fact that both treatments were not explicitly approved for AHA. When a shared decision was reached and patients consented to that treatment, approval for using emicizumab was obtained from the hospital’s administration. After stopping APCC for at least 48 hours (and switching to rhFVIIa in bleeding patients), the first dose of emicizumab was applied subcutaneously (target dose of 3 mg/kg body weight [BW]). Thereafter, rhFVIIa was continued at a lower dose until visible hemostatic response.
Laboratory monitoring was performed with daily APTT, FVIII assays, D-dimer, platelet counts, lactate dehydrogenase (LDH) levels, and organ function parameters to assess for potential adverse events.
Due to the lack of knowledge of emicizumab effects in patients with AHA, further dosing was guided according to clinical bleeding and safety parameters to keep the lowest effective FVIII:h levels (resembling emicizumab plasma levels31,32 ). Emicizumab was therefore continued weekly at 3 mg/kg BW for 2 to 3 additional doses, and dose reduction (to 1.5 mg/kg) and interval prolongation to up to 4 weeks were performed when FVIII:h exceeded 10% (representing approximately emicizumab plasma concentrations of 20 μg/mL).31 At this time, patients were discharged from the hospital and seen every week in the outpatient department to have blood tests and, if FVIII:h was <10%, emicizumab dosing. Lacking better experience, we arbitrarily used FVIII:h >30% to stop emicizumab, and thereafter patients were controlled every 4 weeks. That approach was used so as not to overdose or underdose emicizumab.
Clinical remission was considered when FVIII:h levels exceeded 50%, a level that seems clinically useful in terms of resolved risk of bleeding. Such levels cannot be obtained with emicizumab only26,33 and therefore suggest recovery of patient’s own FVIII. In parallel, complete laboratory remission according to AHA guidelines3,8 was defined as patient’s own FVIII activity (FVIII:b) exceeding 50%.
Assessment of hemostatic efficacy
Hemostatic efficacy was determined according to the recommendations by Tiede et al,34 using hemoglobin levels, need for red blood cell transfusions, hematoma size, or symptoms caused by hematomas as parameters. Insufficient response was defined as the need for a change in hemostatic therapy because of worsening or missing improvement.
Immunosuppressive therapy
In 10 patients, initial immunosuppression was started with a short course of corticosteroids5 (prednisone, 1 mg/kg BW for 1 week, followed by dose tapering over the following 2 weeks). Two patients had uncontrolled diabetes and did not receive steroids. As an attempt to reduce the intensity and adverse effects of immunosuppression, all patients were treated with off-label rituximab2,3,18,20 after consenting to this therapy (in 11 patients within 1 week of diagnosis, 1 patient with a low inhibitor titer started rituximab after 18 weeks because of missing remission).
Statistical methods
Because of the observational nature of this analysis, only descriptive statistical methods were used. The day of the first emicizumab dose was used as the reference day 0. All data are expressed as medians, interquartile ranges (IQRs), and ranges, as indicated; time course was analyzed with the Kaplan-Meier method. IBM SPSS version 24 was used for data analysis and visualization.
Results
Hemostatic treatment summary
Initial hemostatic therapy started 1 day (1-29 days) after initial bleeding (median, range). Three patients were initially treated with APCC (Feiba; Shire) 50 U/kg every 6 hours) and switched to rhFVIIa (Novoseven, Novo-Nordisk; 90 μg/kg every 2 hours) because of insufficient response or adverse effects. Seven patients received initial hemostatic therapy with rhFVIIa. The first dose of emicizumab (= day 0) was given 3 days (1-13 days) after start of initial hemostatic therapy (median dose, 2.7 mg/kg; range, 1.7-3.5 mg/kg). Table 2 summarizes the amount of hemostatic therapy used before and after start of emicizumab, as well as red blood cell transfusions. A median of 5 doses of emicizumab were given (range, 3-9 doses), the last dose after a median of 31 days (range, 12-175 days) (Table 3). One patient needed repetitive small surgery to close her abdominal wound, and was treated prophylactically with additional rhFVIIa injections before and after such surgery.
Hemostatic therapy
| Before emicizumab | After emicizumab | |||
|---|---|---|---|---|
| Median | IQR, range | Median | IQR, range | |
| Hemostatic therapy | ||||
| Duration, d | 3 | 2-8, 0-13 | 1.5 | 0.8-4, 0-16 |
| APCC, ×1000 U | 0 | 0-15, 0-68 | 0 | 0-0, 0-0 |
| rhFVIIa for | ||||
| Active bleeding, mg | 55 | 34-128, 0-402 | 0 | 0-59, 0-224 |
| Prophylaxis, mg | 0 | 0-0, 0-54 | 2.5 | 0-35, 0-275 |
| FVIII concentrate | ||||
| Human, ×1000 U | 0 | 0-5, 0-130 | 0 | 0-0, 0-22 |
| Porcine, ×1000 U | 0 | 0-0, 0-3 | 0 | 0-0, 0-0 |
| RBC concentrates, U | 3 | 0-7, 0-20 | 0.7 | 0-0.5, 0-4 |
| Before emicizumab | After emicizumab | |||
|---|---|---|---|---|
| Median | IQR, range | Median | IQR, range | |
| Hemostatic therapy | ||||
| Duration, d | 3 | 2-8, 0-13 | 1.5 | 0.8-4, 0-16 |
| APCC, ×1000 U | 0 | 0-15, 0-68 | 0 | 0-0, 0-0 |
| rhFVIIa for | ||||
| Active bleeding, mg | 55 | 34-128, 0-402 | 0 | 0-59, 0-224 |
| Prophylaxis, mg | 0 | 0-0, 0-54 | 2.5 | 0-35, 0-275 |
| FVIII concentrate | ||||
| Human, ×1000 U | 0 | 0-5, 0-130 | 0 | 0-0, 0-22 |
| Porcine, ×1000 U | 0 | 0-0, 0-3 | 0 | 0-0, 0-0 |
| RBC concentrates, U | 3 | 0-7, 0-20 | 0.7 | 0-0.5, 0-4 |
Medians, IQR, and range; n = 12.
RBC, red blood cell; U, units.
Emicizumab treatment summary
| Median | IQR | Range | |
|---|---|---|---|
| Start of emicizumab, days after initial bleeding | 8.5 | 2.8-16 | 2-36 |
| Start of emicizumab, days after initial hemostatic therapy | 3 | 2-8 | 1-13 |
| Initial dose of emicizumab, mg/kg | 2.7 | 2.2-3 | 1.7-3.5 |
| Total dose of emicizumab, mg | 907.5 | 694-1148 | 405-1590 |
| No. of doses, n | 5 | 3-7 | 3-9 |
| Last dose of emicizumab, d | 31 | 15-79 | 12-175 |
| Median | IQR | Range | |
|---|---|---|---|
| Start of emicizumab, days after initial bleeding | 8.5 | 2.8-16 | 2-36 |
| Start of emicizumab, days after initial hemostatic therapy | 3 | 2-8 | 1-13 |
| Initial dose of emicizumab, mg/kg | 2.7 | 2.2-3 | 1.7-3.5 |
| Total dose of emicizumab, mg | 907.5 | 694-1148 | 405-1590 |
| No. of doses, n | 5 | 3-7 | 3-9 |
| Last dose of emicizumab, d | 31 | 15-79 | 12-175 |
Medians, IQR, range; n = 12.
Response to emicizumab
As expected, FVIII:C and APTT values normalized within 2 days after the first dose of emicizumab, indicating the well-known artificial effects on these assays. Chromogenic FVIII:h levels exceeded 5% after a median of 4 days (range, 1-13 days) (Table 4). That corresponded with clinically impressive cessation of bleeding. At that time, patient’s FVIII activity, as measured with bovine reagents, was still undetectable. FVIII:h levels exceeded 10% after 11 days (5-13 days); FVIII:b on that day was still <1%. As mentioned, at that point, emicizumab dose was reduced to 1.5 mg/kg BW, and intervals prolonged to every 3 weeks. Emicizumab was stopped as the FVIII:h levels exceeded 30%. Figure 1 illustrates the dynamics of the recovery FVIII of activity after the start of emicizumab.
Response to emicizumab
| Response to emicizumab | Median | IQR | Range |
|---|---|---|---|
| Bleeding stopped, d | 3 | 3-4 | 2-15* |
| Day last rhFVIIa injection | 1.5 | 0.8-4 | 0-16* |
| FVIII (chromogenic), d | |||
| >5% (human) | 4 | 3.3-4.8 | 1-13 |
| >10% (human) | 11 | 7.5-12 | 5-13 |
| >50% (human) | 105 | 33-184 | 9-270 |
| >5% (bovine) | 60 | 26-73 | 12-201 |
| >10% (bovine) | 85 | 33-112 | 12-270 |
| >50% (bovine) | 115 | 67-185 | 18-403 |
| Response to emicizumab | Median | IQR | Range |
|---|---|---|---|
| Bleeding stopped, d | 3 | 3-4 | 2-15* |
| Day last rhFVIIa injection | 1.5 | 0.8-4 | 0-16* |
| FVIII (chromogenic), d | |||
| >5% (human) | 4 | 3.3-4.8 | 1-13 |
| >10% (human) | 11 | 7.5-12 | 5-13 |
| >50% (human) | 105 | 33-184 | 9-270 |
| >5% (bovine) | 60 | 26-73 | 12-201 |
| >10% (bovine) | 85 | 33-112 | 12-270 |
| >50% (bovine) | 115 | 67-185 | 18-403 |
Medians, IQR, range; n = 12.
One patient with an abdominal wound requiring repetitive single doses of rhFVIIa for surgical procedures.
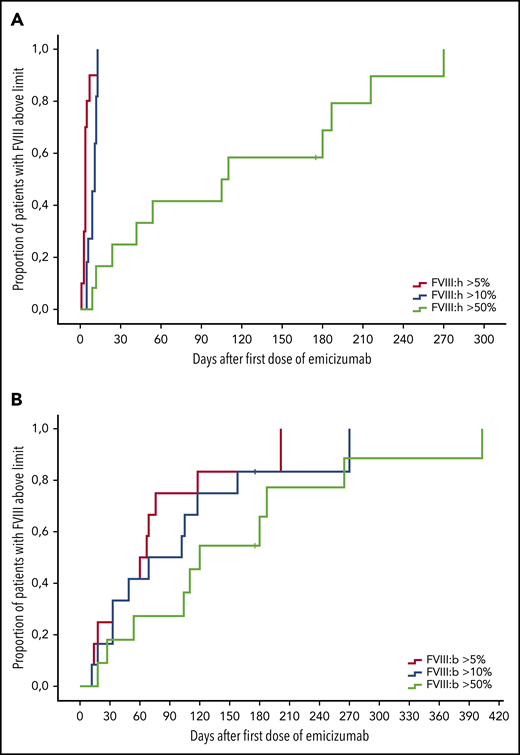
Response of FVIII levels after start of emicizumab therapy. Kaplan-Meier analysis of time to FVIII activity above 5% (blue), 10% (green), or 50% (yellow); n = 12. (A) Chromogenic FVIII assay with human reagents. (B) Chromogenic FVIII assay with bovine reagents. x-axis, Days after first dose of emicizumab. y-axis, Proportion of patients with FVIII levels above threshold.
Response of FVIII levels after start of emicizumab therapy. Kaplan-Meier analysis of time to FVIII activity above 5% (blue), 10% (green), or 50% (yellow); n = 12. (A) Chromogenic FVIII assay with human reagents. (B) Chromogenic FVIII assay with bovine reagents. x-axis, Days after first dose of emicizumab. y-axis, Proportion of patients with FVIII levels above threshold.
Hemostatic efficacy
Even in patients with severe bleeding or surgical wounds, a clinically impressive improvement of bleeding was observed within 3 days (IQR, 3-4 days; range, 2-15 days); 1 patient had mild bleeding associated with small surgical interventions, as mentioned. Also, in the 5 patients with insufficient response to bypassing therapy, bleeding stopped within the first 4 days. No new or breakthrough bleeding events were observed after day 2. Thus, even low emicizumab plasma concentrations seem to protect from bleeding in patients with AHA.
Detection of remission
Figures 2 and 3 show the recovery of FVIII activity levels after the last dose of emicizumab until clinical remission (defined as FVIII:h > 50%). Median time to clinical remission was 105 days (9-270 days). In addition, FVIII activity measured with bovine reagents was helpful to detect remission as defined in the current AHA guidelines, that is, complete recovery of the patient’s FVIII, which occurred after a median of 115 days (range, 18-403 days), corresponding with the data of the GTH-AH 01/2010 trial.16
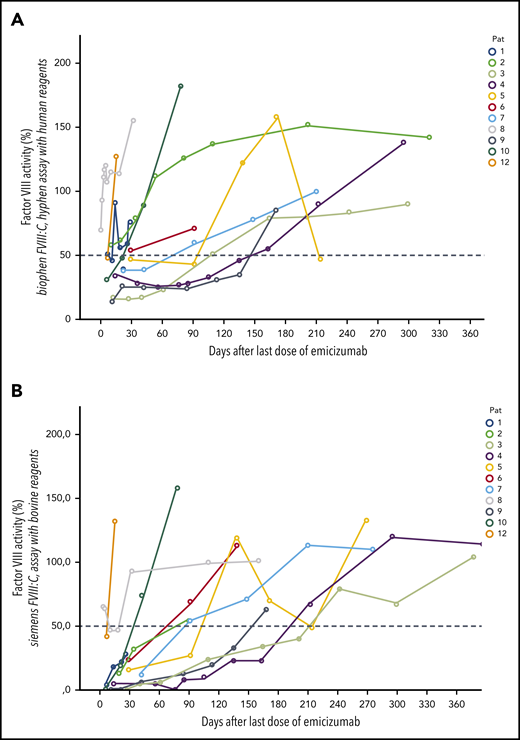
Course of FVIII levels after last dose of emicizumab. FVIII activity levels in 12 patients with AHA after last dose of emicizumab. (A) Chromogenic FVIII assay with human reagents. (B) Chromogenic FVIII assay with bovine reagents.
Course of FVIII levels after last dose of emicizumab. FVIII activity levels in 12 patients with AHA after last dose of emicizumab. (A) Chromogenic FVIII assay with human reagents. (B) Chromogenic FVIII assay with bovine reagents.
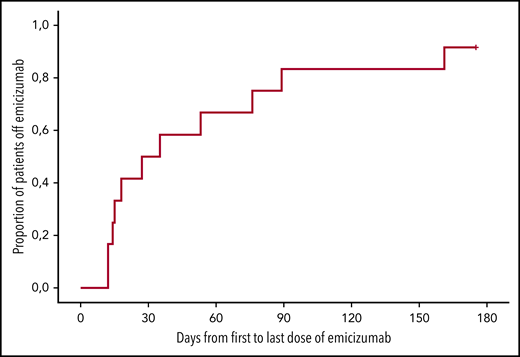
Duration of emicizumab therapy. Kaplan-Meier analysis of the duration of emicizumab therapy (days from first to last dose). n = 12; x-axis, days after first dose of emicizumab.
Duration of emicizumab therapy. Kaplan-Meier analysis of the duration of emicizumab therapy (days from first to last dose). n = 12; x-axis, days after first dose of emicizumab.
Another way to detect remission of the autoimmune process is to measure the concentration of anti-FVIII antibodies. Anti-FVIII IgG antibody concentration declined consecutively after start of immunosuppressive therapy and dropped below 30 AU/mL in 5 patients after a median of 176 days (61-325 days) (Figure 4).
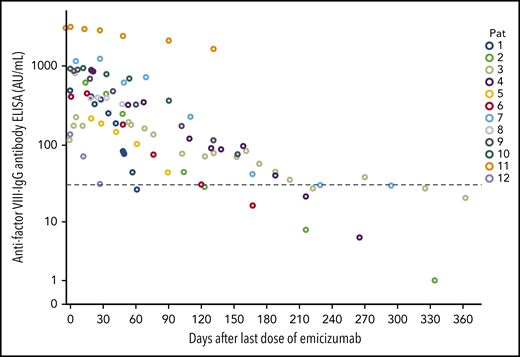
Time course of anti-FVIII IgG antibody concentrations. n = 12; x-axis, days after the first dose of emicizumab; dotted line, 30 AU/mL.
Time course of anti-FVIII IgG antibody concentrations. n = 12; x-axis, days after the first dose of emicizumab; dotted line, 30 AU/mL.
Duration of emicizumab effects in patient’s plasma
Emicizumab has a very long half-life, but detection of low amounts is difficult in patients with AHA, especially when the patient’s own FVIII restores. Conventional emicizumab concentration measurements are based on FVIII assays, either diluted 1-stage clotting assays or chromogenic assays with human reagents, but both are influenced by the patient’s own FVIII. Specific immunologic assays would be necessary to determine the plasma concentrations of emicizumab, but are not commercially available. As an estimation, the ratio of coagulometric FVIII activity using the 1-stage clotting assay (which is very sensitive to emicizumab) to chromogenic FVIII:b activity (which is insensitive to emicizumab) can be helpful, as even trace amounts of emicizumab disturb that ratio. Our data show, that the ratio returned to normal (arbitrarily defined as > 1.5) in 6 patients after a median of 156 days (range, 78-212 days) after the last dose of emicizumab (Figure 5). Thus, in our patients with AHA we can observe a total exposure time to emicizumab of 2354 days (mean, 196 days per patient).
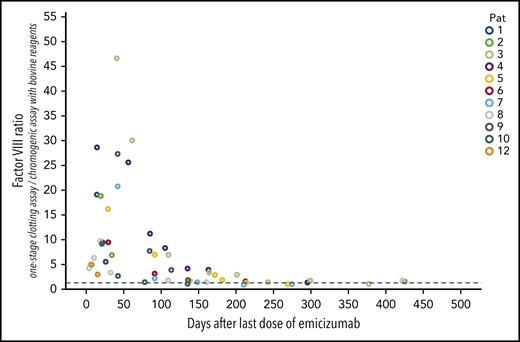
Time course of the ratio of FVIII:C to FVIII:b after the last dose of emicizumab. This ratio was calculated from FVIII activity measured with the 1-stage clotting assay (which is very sensitive to trace amounts of emicizumab; FVIII:C) and the chromogenic assay using bovine reagents (which is insensitive to emicizumab; FVIII:b). n = 12; x-axis, days after the last dose of emicizumab.
Time course of the ratio of FVIII:C to FVIII:b after the last dose of emicizumab. This ratio was calculated from FVIII activity measured with the 1-stage clotting assay (which is very sensitive to trace amounts of emicizumab; FVIII:C) and the chromogenic assay using bovine reagents (which is insensitive to emicizumab; FVIII:b). n = 12; x-axis, days after the last dose of emicizumab.
Immunosuppressive therapy
Steroids were used with care to avoid adverse effects: in 10 patients, initial immunosuppression was started with a short course of corticosteroids (prednisone, 1 mg/kg BW for 1 week, followed by dose tapering over the following 2 weeks). Two patients did not receive steroids because of uncontrolled diabetes or metabolic syndrome. One patient received a short course of cyclophosphamide (8 days, total 800 mg) in addition to steroids. All patients were treated with rituximab as a less toxic, but effective slower immunosuppression (median 4 doses [2-5] and 2500 mg [1000-5000]).
Safety
In a 61-year-old male patient (with a history of severe chronic inflammatory bowel disease for several years), neutropenic sepsis occurred 64 days after start of immunosuppressive therapy with steroids and rituximab, and 20 days after his last dose of emicizumab. He recovered following broad-spectrum antimicrobial therapy and administration of filgrastim. At this time, his FVIII:h level had already exceeded 50%, indicating remission of AHA. One month later (45 days after his last dose of emicizumab), he was admitted to another hospital with a recurrent exacerbation of his chronic inflammatory bowel disease, bowel perforation and peritonitis. The patient rapidly deteriorated and died of this event, which was considered not associated with the applied therapy for AHA, but due to his preexisting chronic bowel disease.
Immunosuppressive therapy was otherwise well tolerated, without any side effects to rituximab or steroids, and no infections or metabolic disturbance in the other 11 patients.
A 79-year-old female patient experienced minor stroke on day 16 on emicizumab during comedication with repetitive single doses of 90 μg/kg rhFVIIa prior to the change of a vacuum assisted closure (VAC) suction system for her large abdominal wound with signs of infection (FVIII:h at this time was 10%, equivalent to emicizumab plasma concentration of about 20 μg/mL,31 and FVIII:b was <1%). She had had inflammation, repetitive surgery with general anesthesia, repetitive rhFVIIa injections, and emicizumab, adiposity, immobilization, and a higher age, as thromboembolic risk factors. rhFVIIa was stopped, emicizumab was continued for 5 more weeks (4 additional doses) after that stroke without worsening of the neurologic situation. No antiplatelet therapy or other anticoagulation was given. She recovered without any sequelae.
Five days before start of emicizumab, an 87-year-old male patient developed non-ST elevation myocardial infarction after 6 days on APCC therapy. He was switched to rhFVIIa for 4 days before emicizumab was started. No anti-platelet therapy was given. He recovered completely.
Emicizumab was otherwise well tolerated, no local or systemic adverse events were observed (Tables 5 and 6). There were no signs of thrombotic microangiopathy, unexplained deviations of laboratory values, or worsening of preexisting clinical conditions. D-dimer levels were elevated in all patients due to bleeding and the underlying conditions, but did not increase further after initiation of emicizumab.
Safety and outcome in patients with AHA treated with emicizumab
| Outcome parameters | Median | IQR | Range |
|---|---|---|---|
| Breakthrough bleedings after FVIII:h >5% | 0 | 0-0 | |
| Day ELISA <30 AU/mL (n = 5) | 176 | 123-216 | 61-325 |
| Days on ICU (n = 1) | 3 | 3-3 | 3-3 |
| Days in hospital | 9.5 | 7-21 | 5-53 |
| Overall survival day 28 | 92% | ||
| Deaths* | 1 (day 82) |
| Outcome parameters | Median | IQR | Range |
|---|---|---|---|
| Breakthrough bleedings after FVIII:h >5% | 0 | 0-0 | |
| Day ELISA <30 AU/mL (n = 5) | 176 | 123-216 | 61-325 |
| Days on ICU (n = 1) | 3 | 3-3 | 3-3 |
| Days in hospital | 9.5 | 7-21 | 5-53 |
| Overall survival day 28 | 92% | ||
| Deaths* | 1 (day 82) |
Medians, IQR, range; n = 12.
ELISA, enzyme-linked immunosorbent assay; ICU, intensive care unit.
Cause of death: peritonitis.
Adverse events after start of emicizumab
| Adverse events | N (day) | Associated with | Outcome |
|---|---|---|---|
| Minor stroke | 1 (16) | Repetitive rhFVIIa injections plus emicizumab, inflammation, repetitive surgery, general anesthesia, adiposity, immobilization, higher age | Recovered |
| Neutropenic sepsis | 1 (55) | Immunosuppression | Recovered |
| Adverse events | N (day) | Associated with | Outcome |
|---|---|---|---|
| Minor stroke | 1 (16) | Repetitive rhFVIIa injections plus emicizumab, inflammation, repetitive surgery, general anesthesia, adiposity, immobilization, higher age | Recovered |
| Neutropenic sepsis | 1 (55) | Immunosuppression | Recovered |
Discussion
Acquired hemophilia is associated with severe bleeding, which can be successfully treated with bypassing agents in most cases. Such treatment, however, is expensive, needs frequent IV injections, and in most cases admission of the patient to experienced centers. In addition, treating physicians tend to apply more intensive immunosuppression to reduce the time to remission to save costs of bypassing therapy, but at the price of more severe, and even lethal, side effects, which was clearly demonstrated in the GTH-AH 01/2010 and other studies.15,16 Although thromboembolic complications with bypassing agents are not frequent,6,9 the transition from low FVIII and compromised hemostasis to rising FVIII and recovering hemostasis considerably increases that risk, and the right time to start pharmacological thromboprophylaxis is hard to determine.8
Emicizumab has been demonstrated to significantly reduce the annualized bleeding rate in hemophiliacs with inhibitors, thus providing an effective prophylaxis. All studies, however, have been conducted in patients with congenital hemophilia. The pharmacological concept of emicizumab, mimicking FVIII as long as its levels are low, but being displaced from binding sites as soon as FVIII levels recover (due to the much higher affinity of FVIII), offers interesting possibilities also in AHA: prevention of spontaneous bleeding, subcutaneous application in long intervals, outpatient management of patients, and no more need for intensive immunosuppression. Our experience from the described cases confirms these thoughts: patients stopped bleeding or did not develop new bleeding, bypassing agents could be stopped, and patients could be discharged early.
There are, however, several possible problems: no clinical studies with emicizumab have been performed in patients with AHA, and formal approval should be seen as valid only for congenital hemophilia. In addition, no females have been treated with emicizumab. Labeling requires 3 consecutive weekly injections with 3 mg/kg to reach the target levels, followed by 1.5 mg/kg per week as maintenance therapy. During the saturation phase the patients would still require bypassing agents to control bleeding. An initial IV injection of emicizumab would overcome that problem, but no IV formulation is currently approved. Pharmacokinetic data from the preclinical trials with emicizumab suggest, that after 1 IV injection of 0.25 mg/kg emicizumab in healthy volunteers the plasma levels of 5.1 μg/mL are reached within 2.5 hours, and decreased with a half-life of 26.7 days.35
However, as demonstrated in our cohort, the hemostatic efficacy was reached already within 3 days after the first subcutaneous injection of emicizumab. Even severe bleeding stopped and bypassing therapy could be rapidly tapered during that period. We did not measure emicizumab plasma concentrations for the mentioned reasons, but chromogenic FVIII:h activity, which has a good correlation with emicizumab plasma levels.31 Indeed, low FVIII:h levels of 2% to 3% became detectable already during the first few days, suggesting that even such low levels (way below the suggested therapeutic range for emicizumab) are hemostatically effective. This is in contrast to the endogenous FVIII, where even levels as high as 30% to 40% do not protect from bleeding because of the complex inhibitor kinetics.36
Other issues arise from the need of special laboratory analysis, as conventional APTT assays and coagulometric FVIII activity assays are oversensitive to emicizumab and normalize even with trace amounts of emicizumab in the patient’s plasma. Such assays can usually not be used to estimate the patient’s bleeding risk. The Bethesda method and all modifications are also not feasible for the quantification of the inhibitor titers in the presence of emicizumab. Chromogenic FVIII activity assays using bovine reagents, in contrast, give reliable results of the patient’s own FVIII levels and inhibitor titers.31 This causes the problem that some important clinical endpoints, like time to effective hemostasis or time of inhibitor eradication, cannot be determined as recommended by current AHA management guidelines.8 Current guidelines use FVIII activity > 50% as definition of remission, as the risk of bleeding is low at such levels.36
We postulate that decisions on dose and intervals of further emicizumab applications is best based on chromogenic FVIII assays using human reagents to keep levels above 10%, but below 30% to avoid potential thromboembolic events in this population with probably higher prothrombotic risk. One possible strategy may be to give an additional dose of 1.5 mg/kg when the level drops below 10%, prolong the intervals if the level ranges between 10 and 30%, and withhold dosing at higher levels.
We do not yet know how to manage patients with breakthrough bleeds or need of surgery under emicizumab as the only hemostatic therapy. No data are available on which emicizumab levels are needed for what type of surgery. Analysis of the HAVEN data, however, suggest that even major surgery is possible in congenital hemophiliacs on emicizumab, without the need for additional factor replacement or bypassing therapy.37
One considerable advantage of emicizumab as a hemostatic agent in AHA is the possibility to use individualized and dose-reduced immunosuppressive therapy and accept a longer time to remission or failure to achieve remission at all. Such a strategy can overcome the well-known high rate of complications due to intensive immunosuppression16 as recommended in the recent AHA guidelines.5 We already followed such an approach and treated with rituximab as a well-tolerated, but slow immunosuppressive method,2,3,18,20,21 and added only a short course of steroids in the first 2 weeks of treatment. Accordingly, only 1 heavily pretreated patient had adverse events to immunosuppression. However, rituximab is not approved for AHA.
The risk of thromboembolic complications in patients with AHA treated with emicizumab is probably low, but still unknown. Although we can observe >2300 exposure days with only 1 arterial event, a prospective trial is necessary to obtain robust safety data. In the meantime, we strongly recommend to avoid combination therapy with bypassing agents whenever possible and to keep attention for thromboembolic events and other adverse reactions.
Our data bear some limitations: this is not a prospective clinical trial, although a prospective approach to collect data were followed. No prospective hemostatic treatment protocol was followed, and an individualized strategy, adapted to the patient’s current situation was used. Emicizumab dosing and laboratory testing, however, was performed according to the described protocol. Another limitation is the fact, that chromogenic FVIII assays using bovine reagents were not yet established in our institution at the time of therapy of the first 6 patients, and these samples were retrospectively re-tested. We feel, however, that chromogenic assays using human reagents are more useful for the clinical management, as they allow not only the detection of upcoming remission, but, more importantly, the individualized dosing of emicizumab.
In conclusion, administration of emicizumab seems to be an important new concept for hemostatic therapy of patients with AHA, with the potential to prevent bleeding, reduce side effects of immunosuppressive therapy, save costs, and protect patients from thromboembolic complications. It may also be beneficial in nonresponding or relapsing patients instead of using third-line immunosuppressive regimens or repeated courses of treatments. Moreover, subcutaneous application of emicizumab in longer intervals offers the possibility of outpatient therapy until remission. Nevertheless, a clinical trial with a sufficient number of patients is necessary to reliably demonstrate these assumptions, and dedicated approval of emicizumab for AHA is necessary.
Data sharing requests may be e-mailed to the corresponding author, Paul Knoebl, at paul.knoebl@muv.ac.at.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors gratefully acknowledge the excellent technical assistance of Silvia Koder, Sabine Belik, Mona Lang, and Yu-Yang Hsiao and their coagulation laboratory staff, and the helpful cooperation of the nursing staff of the hematology ward, intensive care unit, and hemostasis unit of the Medical University of Vienna.
Authorship
Contribution: P.K. had the idea for this project, designed the study, collected data, calculated the statistics, and drafted the manuscript and figures; J.T., W.R.S., and K.G. were responsible for the clinical management of the patients, the documentation of events, and the collection of samples; P.J. and P.Q. were responsible for laboratory testing; and all author proofread the manuscript.
Conflict-of-interest disclosure: P.K. received consultancy honoraria, speaker fees, or travel grants from Novo-Nordisk, Baxalta/Shire/Takeda, CSL Behring, Roche, Sanofi, and Technoclone. J.T. received consultatory honoraria from Daichii Sankyo and Baxalta. P.J. received speaker fees or travel grants from Baxalta/Shire/Takeda. P.Q. received consultancy honoraria or speaker fees from Roche and Baxalta/Shire/Takeda. The remaining authors declare no competing financial interests.
Correspondence: Paul Knoebl, Division for Hematology and Hemostasis, Department of Medicine 1, Medical University of Vienna, Waehringer Guertel 18-20, A-1090 Vienna, Austria; e-mail: paul.knoebl@muv.ac.at.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal