

Key Points
The Emm red cell antigen is carried by the free GPI.
The rare Emm-negative phenotype is associated with developmental disorders.

Abstract
Glycosylphosphatidylinositol (GPI) is a glycolipid that anchors >150 proteins to the cell surface. Pathogenic variants in several genes that participate in GPI biosynthesis cause inherited GPI deficiency disorders. Here, we reported that homozygous null alleles of PIGG, a gene involved in GPI modification, are responsible for the rare Emm-negative blood phenotype. Using a panel of K562 cells defective in both the GPI-transamidase and GPI remodeling pathways, we show that the Emm antigen, whose molecular basis has remained unknown for decades, is carried only by free GPI and that its epitope is composed of the second and third ethanolamine of the GPI backbone. Importantly, we show that the decrease in Emm expression in several inherited GPI deficiency patients is indicative of GPI defects. Overall, our findings establish Emm as a novel blood group system, and they have important implications for understanding the biological function of human free GPI.
Introduction
The Emm-negative rare blood phenotype was first described in 1973 but remains one of the last blood types with an unknown genetic basis. Emm is currently included in the 901 series of high-prevalence red cell antigens. Anti-Emm is rare and seems to be a naturally occurring antibody, as 6 of the reported anti-Emm antibodies were found in nontransfused male subjects.1 The Emm-negative phenotype is believed to be inherited as a recessive trait. The exact nature of the Emm antigen has thus far remained a mystery, but it has been proposed that the Emm antigen is carried on a glycosylphosphatidylinositol (GPI)‐anchored protein (GPI-AP) in the red blood cell (RBC) membrane because its expression is strongly decreased in RBCs from patients with paroxysmal nocturnal hemoglobinuria (PNH).2 PNH is an acquired GPI deficiency caused by somatic mutations in the PIGA gene,3 except in several patients in whom it is caused by combinations of somatic and heterozygous germline mutations in the PIGT gene.4,5 GPI is a glycolipid that tethers >150 different proteins to the cell surface, including the complement-inhibitory glycoprotein (CD59). At least 22 genes termed phosphatidyl inositol glycan (PIG) genes, including PIGA and PIGT, are involved in the synthesis of GPI-APs.6
GPI-AP anchoring is a multistep process that includes synthesis of the GPI precursor in the endoplasmic reticulum (ER), protein attachment to the GPI, and remodeling of the GPI-AP complex in the ER and Golgi (Figure 1).7-9 It is also known that some of the GPI molecules synthesized in the ER are transported to the cell surface and expressed as free, unlinked GPIs.10 To date, germline mutations in PIG/PGAP (post-GPI attachment to proteins) genes reportedly cause various inherited GPI deficiency (IGD) disorders and phenotypes, including multiple congenital anomalies-hypotonia-seizures syndrome (MCAHS) and hyperphosphatasia with mental retardation syndrome/Mabry syndrome.11

Schematic representation of the biosynthesis and trafficking of mammalian GPI-APs. The GPI anchor is synthesized in the ER and transferred to proteins by the GPI-transamidase complex. After protein attachment, GPI moieties are remodeled during transport.
Schematic representation of the biosynthesis and trafficking of mammalian GPI-APs. The GPI anchor is synthesized in the ER and transferred to proteins by the GPI-transamidase complex. After protein attachment, GPI moieties are remodeled during transport.
Here, we show that homozygous null alleles of the PIGG gene cause IGDs and the rare Emm-negative blood phenotype. In addition, we also provide both genetic and cellular evidence that Emm antigen is carried by a free GPI at the cell surface and its epitope is composed of the second and third ethanolamine of the GPI backbone. Finally, we showed that Emm phenotyping of patients with mutations in PIGG, PIGN, and PIGO genes is indicative of GPI defects and could be used in the diagnosis of IGD disorders.
Methods
Subjects
Samples from 3 Emm-negative individuals (P1, P2, and P3), all with anti-Emm, were obtained from a cryopreserved rare reference material collection from the National Reference Center for Blood Group (Paris, France), approved by the local ethics committee (CPP Ile-de-France II) and the French Research Ministry (reference no. DC-2016-2872). Patients with IGD were recruited via PolyWeb and came from the Necker Hospital (Paris, France) and André Mignot Hospital (Versailles, France). PolyWeb is an in-house software system developed by the Bioinformatics Platform of Imagine Institute (https://www.polyweb.fr). The PolyWeb database contains >18 000 in-house exomes.
Clinical descriptions of patients with pathogenic mutations in PIG genes (P1 and patients with IGD) are detailed in supplemental Table 1 (available on the Blood Web site). Informed consent was obtained from both the participants and their legal representatives. RBC samples from voluntary Etablissement Français du Sang blood donors who had consented to the use of their blood for research purposes were used as Emm-positive control subjects.
Serologic testing
Standard hemagglutination tests were used for the assessment of anti-Emm reactivity and RBC typing. The anti-Emm was sourced from 2 unrelated men (P1 and P3) described in 1986 in the original paper.1 By indirect antiglobulin gel testing (ID-Card LISS/Coombs; DiaMed, Bio-Rad, Hercules, CA) at 22°C on papain-treated RBCs, P1, P2, and P3 sera were found to react 3+ (titer 8, score 35), 3+ (titer 128, score 72), and 3+ (titer 8, score 35), respectively. Anti-Emm eluates were prepared after adsorption of human polyclonal anti-Emm from the serum sample of P2 onto a pool of 3 group O papain‐treated RBCs, followed by an acid elution test with the γ ELU‐KIT II device (Immucor Inc, Norcross, GA). The Emm specificity of the eluate was checked by using Emm-positive and Emm-negative RBCs, as well as the absence of contaminating ABO antibodies. The investigation by flow cytometry of the antibody class and subclass of the eluate from P2 was consistent with an immunoglobulin G3 (IgG3) antibody.
Whole-exome sequencing and data analysis
Genomic DNA was extracted from leukocytes by using the MagNA Pure System (Roche Life Science, Penzberg, Germany). Exome capture was performed with the Sure Select Human All Exon Kit (Agilent Technologies, Santa Clara, CA). Agilent Sure Select Human All Exon (58 Mb, V6) libraries were prepared from 3 µg of genomic DNA sheared with an ultrasonicator (Covaris, Woburn, MA) as recommended by the manufacturer. Barcoded exome libraries were pooled and sequenced with a HiSeq2500 system (Illumina Inc, San Diego, CA), generating paired-end reads. After demultiplexing, sequences were mapped on the human genome reference (National Center for Biotechnology Information [NCBI] build 37, hg19 version) with the Burrows-Wheeler Aligner. The mean depth of coverage obtained for the 3 probands’ exome libraries was >120X with ≥96% and ≥94% of the targeted exonic bases covered by at least 15 and 30 independent sequencing reads (≥96% at 15X, ≥94% at 30X). Variant calling was carried out with the Genome Analysis Toolkit (GATK), SAMtools, and Picard tools. Single-nucleotide variants were called with GATK UnifiedGenotyper, whereas insertions and deletions were called with the GATK IndelGenotyper_v2. All variants with a read coverage of 23% and a Phred-scaled quality of 20% were filtered out. All variants were annotated and filtered with PolyWeb, the in-house annotation software program. Pathogenic homozygous or compound heterozygous variants with minor allele frequency <1% (gnomAD database) were considered as potential causes of the Emm-negative phenotype.
Cell culture
K562 cells were cultured in Iscove modified Dulbecco medium, 25 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, and GlutaMAX (Gibco, Carlsbad, CA) supplemented with 10% de-complemented (56°C, 30 minutes) fetal bovine serum, and antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin) at 37°C under a humidified atmosphere containing 5% carbon dioxide. Authentication of our K562 cell line based on genetic characterization was performed by using polymerase chain reaction (PCR) single-locus technology (PowerPlex 21 PCR Kit; Promega, Madison, WI) by Eurofins Genomics (Ebersberg, Germany).
Flow cytometry
Thawed RBCs or K562 cells were washed 3 times in phosphate-buffered saline (PBS) (Gibco) and then resuspended in low-ionic strength buffer supplemented with 0.5% bovine serum albumin and incubated with anti-Emm eluate (1:2) or mouse monoclonal anti-CD59 (1:100; BD Pharmingen, San Diego, CA). Anti-Emm labeling was revealed with anti-human IgG-phycoerythrin (PE) (1:100; Beckman Coulter, Brea, CA), and anti-CD59 labeling was revealed with anti-mouse IgG-PE (1:100; Beckman Coulter). For negative controls of Emm and CD59 staining, cells were incubated with anti-human IgG-PE or anti-mouse IgG-PE only, respectively. In addition, for the negative control of CD59 labeling, similar results were observed by using an isotype primary antibody (mouse IgG). A FACSCanto II flow cytometer (BD Biosciences, Franklin Lakes, NJ) and FlowJo software (FlowJo LLC, Ashland, OR) were used for data acquisition and analysis, respectively. The geometric mean of fluorescence value is indicated for each flow cytometry histogram.
Immunofluorescence confocal microscopy
K562 cells were fixed and permeabilized by using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience/Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions. Briefly, 5 × 106 cells were washed in PBS, incubated in 1X Fix/Perm buffer for 30 minutes at room temperature, and washed twice in 1X Perm buffer (provided in the Foxp3 Transcription Factor Staining Buffer Set). Cells were then incubated with anti-Emm antibody (1:2) in 1X Perm buffer for 120 minutes on ice. The cells were then washed and incubated with Alexa Fluor 488–conjugated goat anti-human IgG (Invitrogen, Paris, France) for 60 minutes on ice. After another set of washes in 1X Perm buffer, samples (0.1 × 106 cells) were cytospun onto glass slides, and 10 µL of ProLong Diamond Antifade Mountant with 4′,6-diamidino-2-phenylindole (Molecular Probes, Eugene, OR) was added before the glass was sealed. Observation was performed by using a 63× objective lens on a Zeiss LSM 700 laser scanning confocal microscope (Carl Zeiss Microscopy GmbH, Jena, Germany).
Western blot
Cell lysates were prepared by homogenization and sonication in sodium dodecyl sulfate (SDS) buffer (Tris-HCl pH 6.8, 5% SDS, 0.2 mM EDTA). Protein quantification was performed by using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Samples were mixed in 2X Tricine-SDS sample buffer, boiled, and separated with Novex 16% Tricine Protein Gels (Thermo Fisher Scientific). Samples were then transferred onto nitrocellulose membranes, blocked with milk for 60 minutes at room temperature, and probed overnight with anti-CD59 (sc-133170, 1/500; Santa Cruz Biotechnology, Dallas, TX), anti-Emm (1:10), or anti-actin (#5125, 1:1000; Cell Signaling Technology, Danvers, MA) diluted in PBS containing 0.1% Tween and 5% bovine serum albumin. Immune complexes were revealed with anti-human IgG horseradish peroxidase (Abliance, Compiègne, France) or anti-mouse horseradish peroxidase (Jackson ImmunoResearch Laboratories, West Grove, PA) using a chemiluminescence kit (Clarity Western ECL Substrate; Bio-Rad).
Immunoprecipitation of CD59
RBC ghosts from Emm-positive and Emm-negative subjects were incubated overnight at 4°C with a mouse monoclonal anti-CD59 antibody (1:50; BD Pharmingen) in PBS/bovine serum albumin. Samples were lysed in lysis buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 0.5% Igepal [MilliporeSigma, Burlington, MA], 0.25% sodium deoxycholic acid, and 0.0025% SDS) for 60 minutes on ice, and the lysates were cleared by centrifugation (15 000g for 15 minutes at 4°C). Immune complexes were purified with Pierce Protein A/G UltraLink Resin (Thermo Fisher Scientific), and the immunoprecipitated proteins were eluted in 2X Tricine-SDS sample buffer at 95°C for 5 minutes and analyzed by western blot as described earlier.
Disruption of PIGA, PIGG, PIGN, PIGO, PIGS, and MPPE1 in K562 by CRISPR/Cas9
Gene editing by CRISPR/Cas9 technology was performed as previously described.12,13 The pSpCas9(BB)-2A-Puro (PX459) expression vector was purchased from Addgene (Cambridge, MA; plasmid #48139). To avoid a potential off-target effect, 3 RNA guides for each of the target genes were selected with CRISPRdirect (http://crispr.dbcls.jp/).14 Oligonucleotides (supplemental Table 1) were ligated into the BbsI linearized PX459 plasmid. The integrity of each cloned guide sequence was checked by Sanger sequencing with PX459 plasmid sequencing primer (5′-GAGGGCCTATTTCCCATGATTCC-3′). The resultant plasmids were purified with NucleoBond Xtra Midi Plus EF (Macherey-Nagel, Düren, Germany). Five micrograms of plasmid were electroporated with 106 K562 cells using Nucleofector II (kit V-program T-016; Lonza, Basel, Switzerland). Cells were seeded at a density of 150 000 cells/mL 48 hours before electroporation. Viability was >95% on the day of transfection. Transfected K562 cells were grown with puromycin (3-5 μg/mL during 4 days) in the culture medium 24 hours after transfection until selection were done. Cells were then grown without puromycin. Generation of INDEL events in the targeted exon was assessed by using T7 endonuclease I enzyme (New England Biolabs, Ipswich, MA) (supplemental Figure 2). Knockout (KO) transfectant cell populations were determined by flow cytometry analysis of Emm (for PIGG KO) and CD59 (PIGA, PIGS, PIGN, and PIGO KO) expression (supplemental Figure 3). After fluorescence-activated cell sorting, KO cells were confirmed by sequencing the target sites (supplemental Figure 4), TIDE analyzing (https://tide.nki.nl/)15 (supplemental Figure 5), and quantitative reverse transcription-PCR (qRT-PCR) (supplemental Figure 6). The K562 wild-type (WT) cells correspond to K562 cells transfected with Cas9 nuclease alone without RNA-guide.
RNA extraction, complementary DNA synthesis, and RT-qPCR
K562 KO cells were collected by centrifugation, and total RNA was extracted by using TRIzol (Invitrogen, Paris, France). Complementary DNA (cDNA) synthesis was performed by using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. ABsolute Blue qPCR SYBR Green Mix (Thermo Fisher Scientific) was then used to amplify specific cDNA fragments in the CFX96 Real Time System (Bio-Rad) using primers listed in supplemental Table 2.
Plasmid construction and cell transfection
Human PIGG WT cDNA was obtained in the pcDNA3.1(+)-C-eGFP vector (provided by GenScript) and subcloned into the pCEP4 vector (Invitrogen). To obtain PIGG H216Y mutant cDNA, an Agilent QuikChange site-directed mutagenesis kit was used according to the manufacturer’s instructions. Plasmid sequences were confirmed before cell transfection by Sanger sequencing. K562 PIGG KO cells were transfected with 5 µg of pCEP4-Empty, pCEP4-hPIGGWT, or pCEP4-hPIGGH214Y using Amaxa Cell Line Nucleofector Kit V (Lonza) according to the manufacturer’s instructions. Stable transfectants were obtained after 10 days of selection with hygromycin B (0.2 mg/mL; Invitrogen).
Lipid adsorption
Thawed RBCs from Emm-positive and Emm-negative subjects were incubated for 120 minutes with 0.5 mg of porcine brain polar lipid extract (Avanti Polar Lipids, Alabaster, AL) in PBS containing 1% methanol. After 3 washes in PBS, the surface level of the Emm antigen was analyzed by flow cytometry.
Results
PIGG underlies the Emm blood group
To identify the molecular basis of the Emm blood group antigen, whole-exome sequencing was performed in genomic DNA from 3 unrelated Emm-negative non-PNH probands (P1, P2, and P3). Variant-filtering strategies led to the identification of 3 mutations in the common gene PIGG (GenBank: NM_001127178). Proband 1 carried a homozygous variant, c.640C>T; (p. His214Tyr) (Figure 2A), which is absent from public and in-house databases (Imagine Institute, PolyWeb). Proband 2 was homozygous for a G deletion at the +1 position of the splice donor site of intron 5 (c.901+1delG) (Figure 2B). The splice mutation has been reported in gnomAD only in the heterozygous state, with an allele frequency of 9/248268. Proband 3 carries a 4 kb deletion that removed exon 6 (Figure 2C). PIGG encodes a 983 amino acid protein, a GPI ethanolamine phosphate (EtNP) transferase 2 (also known as phosphatidylinositol glycan anchor biosynthesis, class G), that is involved in the addition of a side-chain modification on the second mannose of GPI.16 Flow cytometry analysis of Emm-negative RBCs with a specific anti-Emm antibody eluate confirmed the absence of the Emm antigen in PIGG-mutated RBCs, whereas GPI-AP CD59 was normally expressed (Figure 2D).
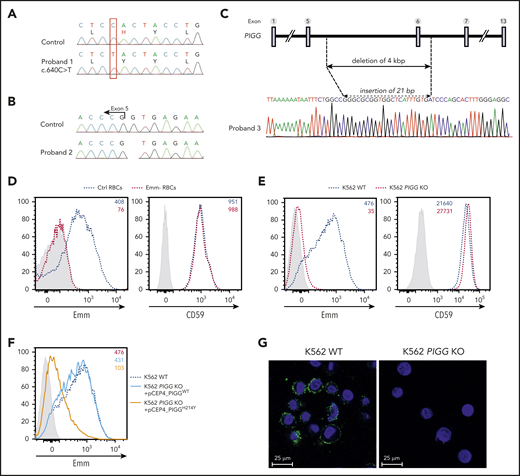
Mutations in the PIGG gene are responsible for the Emm-negative blood group. (A-B) Sanger sequencing of PIGG mutations found in Emm-negative proband 1 and proband 2, respectively. (C) Schematic representation of the deletion in the PIGG gene and Sanger sequencing of Emm-negative proband 3. (D) Cell surface expression of the Emm antigen and CD59 in control RBCs (Ctrl RBCs) and Emm-negative RBCs (Emm- RBCs). The gray profile corresponds to RBCs incubated with only the secondary antibody. (E) Cell surface expression of the Emm antigen and CD59 in K562 WT and K562 PIGG KO cells. The gray profile corresponds to K562 cells incubated with only the secondary antibody. (F) Surface level of Emm antigen in K562 PIGG KO cells stably transfected with WT PIGG cDNA or mutant PIGG cDNA (c.640C>T; p.H214Y). The gray profile corresponds to K562 cells incubated with only the secondary antibody. (G) Immunofluorescence analysis of Emm antigen shows the plasma membrane localization of this antigen. Nonpermeabilized WT and PIGG KO K562 cells were stained by using an anti-Emm (green) antibody and 4′,6-diamidino-2-phenylindole (blue).
Mutations in the PIGG gene are responsible for the Emm-negative blood group. (A-B) Sanger sequencing of PIGG mutations found in Emm-negative proband 1 and proband 2, respectively. (C) Schematic representation of the deletion in the PIGG gene and Sanger sequencing of Emm-negative proband 3. (D) Cell surface expression of the Emm antigen and CD59 in control RBCs (Ctrl RBCs) and Emm-negative RBCs (Emm- RBCs). The gray profile corresponds to RBCs incubated with only the secondary antibody. (E) Cell surface expression of the Emm antigen and CD59 in K562 WT and K562 PIGG KO cells. The gray profile corresponds to K562 cells incubated with only the secondary antibody. (F) Surface level of Emm antigen in K562 PIGG KO cells stably transfected with WT PIGG cDNA or mutant PIGG cDNA (c.640C>T; p.H214Y). The gray profile corresponds to K562 cells incubated with only the secondary antibody. (G) Immunofluorescence analysis of Emm antigen shows the plasma membrane localization of this antigen. Nonpermeabilized WT and PIGG KO K562 cells were stained by using an anti-Emm (green) antibody and 4′,6-diamidino-2-phenylindole (blue).
Next, to validate that Emm expression is controlled by PIGG, the CRISPR/Cas9 approach was used to inactivate this gene in K562 cells. Notably, flow cytometry analysis revealed a strong decrease in Emm expression in PIGG KO cells but normal expression of CD59 (Figure 2E). In addition, PIGG cDNA bearing the p.His214Tyr variant found in the P1 proband could not rescue the surface abundance of Emm in these cells, whereas the overexpression of WT PIGG could (Figure 2F). Although GPI biosynthesis is carried out on the ER membrane, immunofluorescence staining of nonpermeabilized WT and PIGG-deficient cells confirmed the cell surface expression of the Emm antigen (Figure 2G). These results show that the PIGG gene underlies the Emm blood group antigen.
Emm antigen corresponds to free GPIs
Because the Emm antigen is not expressed in PNH type III cells, which are caused by somatic null mutations in the PIGA gene,2 we examined Emm expression in PIGA-deficient K562 cells. As expected, these cells lost CD59 and the Emm antigen (Figure 3A), confirming that Emm expression is related to GPI. Next, to address whether the Emm antigen is carried by the GPI backbone or by a protein attached to GPI, we inactivated PIGS, a major protein of the GPI-transamidase complex responsible for GPI attachment to proteins.17 Interestingly, flow cytometric analysis showed that knockout of PIGS in K562 cells greatly increased Emm expression and, as expected, abolished CD59 expression (Figure 3B). Accordingly, immunofluorescence staining of permeabilized cells showed that the subcellular localization of Emm was shifted to the cell surface after PIGS inactivation (Figure 3C), which indicates that Emm is not carried by a protein attached to GPI.
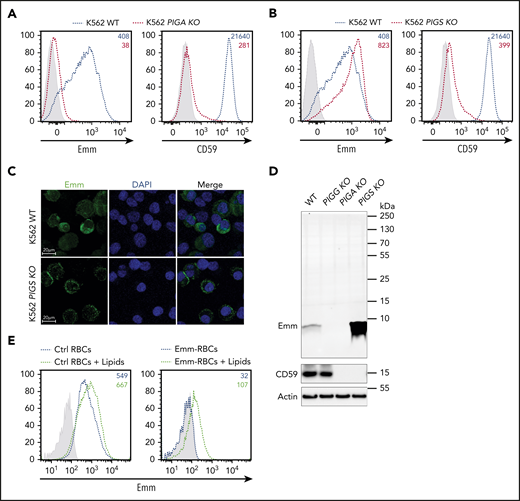
Emm antigen is carried by the free GPI. (A) Cell surface expression of the Emm antigen and CD59 in K562 WT and K562 PIGA KO cells. The gray profile corresponds to K562 cells incubated with only the secondary antibody. (B) Cell surface expression of the Emm antigen and CD59 in K562 WT and K562 PIGS KO cells. The gray profile corresponds to K562 cells incubated with only the secondary antibody. (C) Immunofluorescence analysis of the Emm antigen. Fixed and permeabilized cells were stained with an anti-Emm antibody (green) and 4′,6-diamidino-2-phenylindole (DAPI; blue). (D) Western blot analysis of Emm and CD59 expression in K562 WT, K562 PIGA KO, and K562 PIGS KO cells. Twenty micrograms of protein from cell lysates was resolved by SDS–polyacrylamide gel electrophoresis under reducing conditions and probed with anti-Emm and anti-CD59 antibodies. Actin was used as a loading control. (E) Surface level analysis of Emm antigen on RBCs after incubation with brain polar lipids. Control RBCs (left panel) or Emm-negative RBCs (right panel) were incubated in 1% methanol in PBS containing 0.5 mg of brain polar lipids (black dotted lines) or with only 1% methanol in PBS (blue dotted lines) for 120 minutes. Emm antigen adsorption on the RBC surface was monitored by fluorescence-activated cell sorting using the anti-Emm antibody.
Emm antigen is carried by the free GPI. (A) Cell surface expression of the Emm antigen and CD59 in K562 WT and K562 PIGA KO cells. The gray profile corresponds to K562 cells incubated with only the secondary antibody. (B) Cell surface expression of the Emm antigen and CD59 in K562 WT and K562 PIGS KO cells. The gray profile corresponds to K562 cells incubated with only the secondary antibody. (C) Immunofluorescence analysis of the Emm antigen. Fixed and permeabilized cells were stained with an anti-Emm antibody (green) and 4′,6-diamidino-2-phenylindole (DAPI; blue). (D) Western blot analysis of Emm and CD59 expression in K562 WT, K562 PIGA KO, and K562 PIGS KO cells. Twenty micrograms of protein from cell lysates was resolved by SDS–polyacrylamide gel electrophoresis under reducing conditions and probed with anti-Emm and anti-CD59 antibodies. Actin was used as a loading control. (E) Surface level analysis of Emm antigen on RBCs after incubation with brain polar lipids. Control RBCs (left panel) or Emm-negative RBCs (right panel) were incubated in 1% methanol in PBS containing 0.5 mg of brain polar lipids (black dotted lines) or with only 1% methanol in PBS (blue dotted lines) for 120 minutes. Emm antigen adsorption on the RBC surface was monitored by fluorescence-activated cell sorting using the anti-Emm antibody.
To confirm this finding, we then used an anti-Emm antibody for western blot analysis of the generated KO K562 cell lines. Interestingly, no specific band corresponding to GPI-AP was detected in the 250-to-10 kDa region; however, a specific band appeared at 5 kDa in WT K562 cells but not in PIGG and PIGA KO cells (Figure 3D; supplemental Figure 1). In accordance with the flow cytometry data, the intensity of the Emm band was enhanced in PIGS KO cells. These results indicate that the Emm antigen is carried by unlinked GPI (free GPI), which was further supported by the increase in Emm labeling of Emm-negative and control RBCs upon incubation with polar lipid extracts presumably containing unlinked GPI glycolipid (Figure 3E). In addition, because PIGG underlies Emm expression, we conclude that the second EtNP of free GPIs is a part of the Emm epitope. However, this EtNP transferred by PIGG was removed by the phosphodiesterase PGAP5 (encoded by the MPPE1 gene) after the attachment of GPI to proteins and is not present in the most mature GPI-APs (Figure 1).9
To determine whether the presence of EtNP2 in GPI-AP leads to an increase in Emm expression (Figure 4A), we generated PGAP5-deficient cells (MPPE1 KO cells) and analyzed them with anti-Emm. Flow cytometry and immunofluorescence analysis showed that PGAP5 inactivation does not increase the expression of Emm, although GPI-AP is normally expressed (Figure 4B-C). Normal expression of CD59 and Emm was confirmed by western blot analysis, which also indicates that the migration profile of the Emm band remains unchanged (Figure 4D). Finally, immunoprecipitation of CD59 from Emm-negative and control RBCs was performed. As expected, the Emm antigen was not coprecipitated with CD59 from Emm-positive RBCs (Figure 4E). All these data showed that anti-Emm does not recognize the second EtNP in GPI-APs and confirmed that the Emm antigen is carried by free GPI.
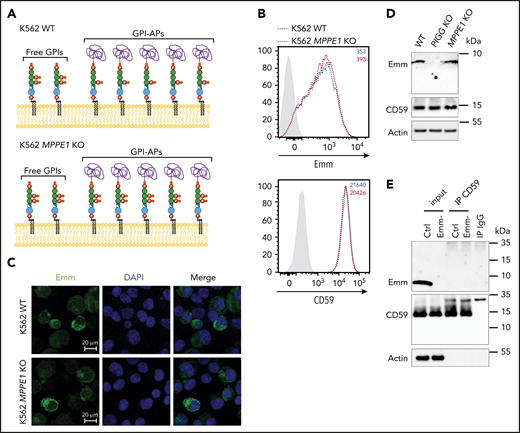
Anti-Emm did not recognize the second EtNP on the GPI-APs. (A) Schematic representation of the free GPI and GPI-AP structures in K562 WT and K562 PGAP5-deficient cells (MPPE1 KO) cells. (B) Cell surface expression of the Emm antigen and CD59 in K562 WT and K562 MPPE1 KO cells. The gray profile corresponds to K562 cells incubated with only the secondary antibody. (C) Immunofluorescence analysis of Emm antigen. Fixed and permeabilized cells were stained with the anti-Emm antibody and 4′,6-diamidino-2-phenylindole (DAPI). (D) Western blot analysis of Emm and CD59 expression in K562 WT, K562 PIGG KO, and K562 MPPE1 KO cells. Twenty micrograms of protein from cell lysates was resolved by SDS–polyacrylamide gel electrophoresis under reducing conditions and probed with anti-Emm and anti-CD59 antibodies. Actin was used as a loading control. (E) CD59 was immunoprecipitated from the control RBC membrane (Ctrl) or Emm-negative RBC membrane (Emm-). Input and immune complexes (IP CD59) were analyzed by SDS–polyacrylamide gel electrophoresis under reducing conditions with heat denaturation, followed by western blot analyses using anti-Emm and anti-CD59 antibodies. Actin was used as a loading control.
Anti-Emm did not recognize the second EtNP on the GPI-APs. (A) Schematic representation of the free GPI and GPI-AP structures in K562 WT and K562 PGAP5-deficient cells (MPPE1 KO) cells. (B) Cell surface expression of the Emm antigen and CD59 in K562 WT and K562 MPPE1 KO cells. The gray profile corresponds to K562 cells incubated with only the secondary antibody. (C) Immunofluorescence analysis of Emm antigen. Fixed and permeabilized cells were stained with the anti-Emm antibody and 4′,6-diamidino-2-phenylindole (DAPI). (D) Western blot analysis of Emm and CD59 expression in K562 WT, K562 PIGG KO, and K562 MPPE1 KO cells. Twenty micrograms of protein from cell lysates was resolved by SDS–polyacrylamide gel electrophoresis under reducing conditions and probed with anti-Emm and anti-CD59 antibodies. Actin was used as a loading control. (E) CD59 was immunoprecipitated from the control RBC membrane (Ctrl) or Emm-negative RBC membrane (Emm-). Input and immune complexes (IP CD59) were analyzed by SDS–polyacrylamide gel electrophoresis under reducing conditions with heat denaturation, followed by western blot analyses using anti-Emm and anti-CD59 antibodies. Actin was used as a loading control.
The Emm epitope is composed of the second and third EtNPs of the free GPI
The core backbone of GPI is formed by three EtNPs, three mannoses (Man), one non–N-acetylated glucosamine, and inositol phospholipids. Each mannose is modified by one EtNP group. The transfer of EtNPs to Man1, Man2, and Man3 is catalyzed by three EtNP transferases: PIGN, PIGG, and PIGO, respectively (Figure 5A).16,18,19 To determine if EtNP1 and/or EtNP3, in addition to EtNP2, are also parts of the free GPI epitope recognized by anti-Emm, PIGN and PIGO KO cells were generated. Flow cytometric analysis showed that knockout of PIGN in WT K562 cells weakly increased cell surface anti-Emm staining, whereas inactivation of PIGO completely abolished the expression of Emm (Figure 5B-C). Flow cytometry results were confirmed by western blot analysis of the cell lysates with anti-Emm (Figure 5D). Overall, we show that the Emm epitope is composed of the second and third EtNPs of the free GPI.

The Emm antigen is composed of the second and third EtNPs of the free GPI. (A) Schematic representation of the free GPI in the plasma membrane. PIGN, PIGG, and PIGO catalyze the addition of EtNPs on the first, second, and third mannose, respectively. (B) Cell surface expression of the Emm antigen in K562 WT and K562 PIGN KO cells. The gray profile corresponds to K562 cells incubated with only the secondary antibody. (C) Cell surface expression of the Emm antigen in K562 WT and K562 PIGO KO cells. The gray profile corresponds to K562 cells incubated with only the secondary antibody. (D) Western blot analysis of Emm and CD59 expression in K562 WT, K562 PIGG KO, K562 PIGA KO, K562 PIGN KO, and K562 PIGO KO cells. Twenty micrograms of protein from cell lysates was resolved by SDS–polyacrylamide gel electrophoresis under reducing conditions and probed with anti-Emm and anti-CD59 antibodies. Actin was used as a loading control.
The Emm antigen is composed of the second and third EtNPs of the free GPI. (A) Schematic representation of the free GPI in the plasma membrane. PIGN, PIGG, and PIGO catalyze the addition of EtNPs on the first, second, and third mannose, respectively. (B) Cell surface expression of the Emm antigen in K562 WT and K562 PIGN KO cells. The gray profile corresponds to K562 cells incubated with only the secondary antibody. (C) Cell surface expression of the Emm antigen in K562 WT and K562 PIGO KO cells. The gray profile corresponds to K562 cells incubated with only the secondary antibody. (D) Western blot analysis of Emm and CD59 expression in K562 WT, K562 PIGG KO, K562 PIGA KO, K562 PIGN KO, and K562 PIGO KO cells. Twenty micrograms of protein from cell lysates was resolved by SDS–polyacrylamide gel electrophoresis under reducing conditions and probed with anti-Emm and anti-CD59 antibodies. Actin was used as a loading control.
The surface expression of Emm antigen is altered in RBCs from patients with IGD
Importantly, the expression level of the Emm antigen is under the control of several genes involved in GPI biosynthesis, suggesting that loss-of-function mutations in PIG genes could be associated with a weak Emm blood phenotype. This point is very important for analyzing cells from patients with IGD caused by germline mutations in at least 21 PIG genes. In IGD patients, the expression level of GPI-APs in blood cells was often not indicative of the gene defect and did not correlate with the severity of the clinical phenotype.20 This finding is confirmed by the normal expression of CD59 (Figure 6) in RBCs from the P1 proband even though the subject had an intellectual disability and hypotonia,1 potentially caused by the pathogenic His214Tyr mutation in PIGG (supplemental Table 1). By contrast, the Emm-negative phenotype indicated a defect in GPI biosynthesis. Thus, we decided to investigate whether pathogenic mutations in other PIG genes involved in IGDs are associated with low expression of the Emm antigen on the RBC surface. Patients with IGD were clinically investigated at the Necker Hospital in Paris and selected from the Exome database of the Imagine Institute via the PolyWeb interface. The patient with PIGN was a female who died suddenly of a severe epileptic seizure at the age of 4 years. She had a global developmental delay and intractable epilepsy, severe hypotonia, and muscular atrophy. Exome sequencing identified a pathogenic mutation c.284G > A (p. Arg95Gln) in the PIGN gene. This mutation has been described previously in patients with MCAHS.21 Flow cytometry analysis of her RBCs showed a normal expression level of CD59 and a slight increase in Emm expression compared with that of control RBCs. The increase of Emm expression in these RBCs is consistent with PIGN-deficient K562 cell line (Figure 5B). The patient with PIGO is a 10-year-old son of Egyptian consanguineous parents. He has intellectual disability, cerebral atrophy, hypotonia, and delayed psychomotor development (supplemental Table 3). The c.23T>C (p. Leu8Pro) variant in PIGO has been reported in another patient with IGD (ClinVar database, rs755191263). Interestingly, flow cytometry analysis of RBCs from this patient showed a strong decrease in Emm expression, whereas CD59 expression was marginally reduced compared with that of an unrelated control. Finally, the PIGA patient is a 6-year-old boy who has refractory epilepsy and a severe developmental delay, including delays in language development and motor ability; he is bedridden and does not make eye contact. This patient carries a maternal pathogenic mutation, c.241C>T (p.Arg81Cys), which was reported in another patient with MCAHS.22 Flow cytometry analysis showed that the germline mutation c.241C>T in PIGA does not severely affect Emm and CD59 expression in mature RBCs, although this mutation is responsible for the aberrant development of this patient. Collectively, the clinical features resulting from pathogenic variants in PIGG and PIGO genes were corroborated by the altered expression of Emm in RBCs, whereas GPI-AP expression was not altered.
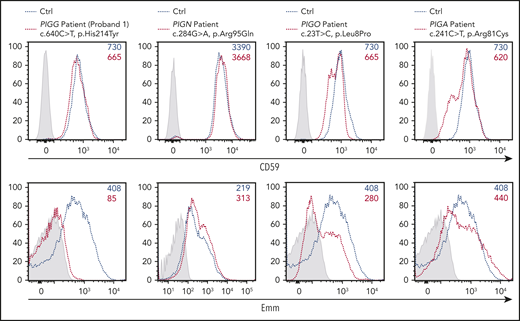
Expression of the Emm antigen vs CD59 in RBCs from IGD patients. Cell surface expression of CD59 (top panel) and the Emm antigen (bottom panel) in RBCs from PIGG (proband 1), PIGA, PIGN, and PIGO patients. The gray profile corresponds to RBCs incubated with only the PE-conjugated anti-mouse (top panel) and anti-human (bottom panel) IgG antibodies.
Expression of the Emm antigen vs CD59 in RBCs from IGD patients. Cell surface expression of CD59 (top panel) and the Emm antigen (bottom panel) in RBCs from PIGG (proband 1), PIGA, PIGN, and PIGO patients. The gray profile corresponds to RBCs incubated with only the PE-conjugated anti-mouse (top panel) and anti-human (bottom panel) IgG antibodies.
Discussion
The major finding in this study is that free, unlinked GPI is expressed at the RBC surface and carries the Emm blood antigen. To date, the GPI is involved in the expression of 6 other blood group systems: YT, DO, CROM, JMH, CD59, and KANNO.2,23,24 However, Emm is the only antigen carried by the GPI backbone and not by a protein linked to GPI. Although the GPI is synthesized in the ER, the presence of several GPI precursors at the cell surface has been documented in mammalian cells and parasites such as Toxoplasma gondii and Plasmodium falciparum.25,26 Plasmodium-free GPI is highly immunogenic and elicits a parasite-specific IgG response in people living in malaria-endemic areas.26 Consistently, our newly characterized anti-free GPI, anti-Emm, was often described as a naturally occurring antibody in all Emm-negative patients. In addition, although anti-Emm recognizes the GPI precursor that contains three EtNPs, only the second and third EtNP are included in the epitope. The second EtNP, transferred by PIGG, is a transient side-chain that is removed by PGAP5 and is absent from the mature GPI-APs (Figure 1).9 The normal surface expression of Emm in PGAP5-defective cells is consistent with the fact that anti-Emm is specific for free GPI. Because Emm is also expressed in platelet and granulocyte surfaces (supplemental Figure 7), we provide the unequivocal demonstration that free GPIs are expressed in various human blood cells and are normal components of cell membranes.
Although almost nothing is known about the physiological roles of the free GPIs in human cells, we have recently reported a pathologic effect of the free GPIs in patients with PNH that have PIGT mutations.5 PIGT deficiency results on an abnormal accumulation of the unlinked free GPIs that enhances the activation of NLRP3 inflammasome and complement. Although both PIGA-PNH and PIGT-PNH are characterized by GPI-AP deficiency, they display different molecular alterations. As with PIGS, PIGT is a GPI transamidase involved in the attachment of GPI to proteins in the ER.17 PIGA is required for the first step in GPI biosynthesis; therefore, no GPI precursor is generated in PIGA-defective cells (Figure 1). Consistently, the Emm antigen (free GPI) is highly expressed in PIGS KO cells (Figure 3B) but is absent in PIGA-PNH RBCs.2 Overall, anti-Emm seems to be a new helpful tool for detecting GPI defect, and Emm phenotyping could be associated with GPI-AP analysis for the diagnosis of PNH disease.
Defective biosynthesis of the GPI is caused by several different genetic mechanisms. In PNH disease, the GPI deficiency is caused by somatic null or nearly null mutations in PIGA that occur in the hematopoietic stem cells. By contrast, germline mutations in PIGA and other PIG genes cause IGD.27 Null germline mutations in PIGA are believed to be embryonic lethal,28 suggesting that p.Arg81Cys found in our patient has residual function (supplemental Table 1). Consistent with previous findings,29 CD59 expression in RBCs from our PIGA-IGD patient is not strongly affected, which is in accordance with the absence of intravascular hemolysis and erythroid disorders. The GPI-AP defect in patients with IGD is often more conspicuous on granulocytes than in erythrocytes, suggesting that the consequence of germline mutations in PIG genes is tissue specific.
In line with this finding, many differences in GPI structure were found among different mammalian tissues and cell lines, suggesting that the GPI anchor may be regulated in a tissue-specific way. In fact, the most prominent clinical symptoms of IGD are neurologic ones, including seizures, developmental delay/intellectual disability, cerebral atrophy, and hypotonia.11 Presumably, the partial reduction of GPI-APs is less tolerated in neuronal development than hematopoiesis, as shown in a human-induced pluripotent stem cell model.30 Conversely, the homozygous null alleles of PIGG found in Emm-negative patients do not cause embryonic lethality but do cause IGD (supplemental Table 1). Interestingly, these mutations result in total absence of a free GPI with unaltered GPI-AP expression in both RBCs and PIGG KO cells (Figure 2D-E). In addition, we showed that the GPI defect in the patient with PIGO-IGD is also detected by the altered expression of Emm in RBCs, whereas GPI-AP expression was not altered. These findings are consistent with the implication of both PIGG and PIGO genes in the epitope structure of Emm (Figure 5). Further analyses in patients with IGD that have other PIG mutations are needed to validate the relevance of Emm expression at RBC surface in this pathology.
In summary, we provide both genetic and cellular evidence that the Emm antigen is carried by free GPI at the RBC surface and establish Emm as a novel human blood group system. The natural occurrence of anti-Emm is consistent with the high immunogenicity of free GPI reported in numerous microorganisms31,32 and suggests a potential role for this antibody in protection against some infectious agents. These findings have important implications for understanding the biological function of human free GPI and the intracellular trafficking of GPI. A pilot study in patients with IGD and PNH would be necessary to evaluate the clinical relevance of an Emm phenotyping approach compared with GPI-AP expression studies.
For original data, please contact the corresponding author (slim.azouzi@inserm.fr).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
Acknowledgments
The authors thank the Emm probands and PIG patients and their families for their participation in the study. They also thank Jean-Philippe Semblat for technical help with confocal microscopy and all members of the Centre National de Référence pour les Groupes Sanguins for the management of blood samples.
This work was supported by INSERM, l’Association Recherche et Transfusion (ART 2017), the Institut National de la Transfusion Sanguine, and the Laboratory of Excellence GR-Ex (ANR-11-LABX-0051). The Labex GR-Ex is funded by the IdEx Program “Investissements d’Avenir” of the French National Research Agency (ANR-18-IDEX-0001).
Authorship
Contribution: J.-P.C., Y.C., C.L.V.K., O.H., T.P., and S.A. conceived and designed the study; R.D., G.N., A.W., Y.M., M.M., C.V., N.C., G.B., and S.A. performed the experiments; R.D., A.W., and G.N. designed and performed the CRISPR/Cas-9 experiments; R.D., G.N., A.W., Y.M., C.M., G.B., V.C., T.K., and S.A. analyzed the data; H.M., S.W., B.K., M.H., K.S., N.C., A.R., S.L., V.C., and O.H. analyzed data related to IGD patients; S.A. wrote the manuscript with editing assistance from R.D., Y.C., C.L.V.K., J.-P.C., T.K., and T.P; and all authors approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Slim Azouzi, INSERM, UMR_S1134, INTS, 6 Rue Alexandre Cabanel, 75015 Paris, France; e-mail: slim.azouzi@inserm.fr; Thierry Peyrard, INSERM, UMR_S1134, INTS, 6 Rue Alexandre Cabanel, 75015 Paris, France; e-mail: tpeyrard@ints.fr; or Taroh Kinoshita, Yabumoto Department of Intractable Disease Research, Research Institute for Microbial Diseases, Osaka University, 3-1 Yamada-oka, Suita, Osaka 565-0871, Japan; e-mail: tkinoshi@biken.osaka-u.ac.jp.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal