

Key Points
Acalabrutinib demonstrated safety and durable remission in a phase 1/2 clinical trial in previously untreated CLL.
Long-term data from this study support use of acalabrutinib as initial therapy for patients with symptomatic CLL.

Abstract
Acalabrutinib has demonstrated significant efficacy and safety in relapsed chronic lymphocytic leukemia (CLL). Efficacy and safety of acalabrutinib monotherapy were evaluated in a treatment-naive CLL cohort of a single-arm phase 1/2 trial (ACE-CL-001). Adults were eligible for enrollment if chemotherapy was declined or deemed inappropriate due to comorbidities (N = 99). Patients had a median age of 64 years and 47% had Rai stage III/IV disease. Acalabrutinib was administered orally 200 mg once daily, or 100 mg twice daily until progression or intolerance. A total of 99 patients were treated; 57 (62%) had unmutated immunoglobulin heavy-chain variable gene, and 12 (18%) had TP53 aberrations. After median follow-up of 53 months, 85 patients remain on treatment; 14 discontinued treatment, mostly because of adverse events (AEs) (n = 6) or disease progression (n = 3). Overall response rate was 97% (90% partial response; 7% complete response), with similar outcomes among all prognostic subgroups. Because of improved trough BTK occupancy with twice-daily dosing, all patients were transitioned to 100 mg twice daily. Median duration of response (DOR) was not reached; 48-month DOR rate was 97% (95% confidence interval, 90-99). Serious AEs were reported in 38 patients (38%). AEs required discontinuation in 6 patients (6%) because of second primary cancers (n = 4) and infection (n = 2). Grade ≥3 events of special interest included infection (15%), hypertension (11%), bleeding events (3%), and atrial fibrillation (2%). Durable efficacy and long-term safety of acalabrutinib in this trial support its use in clinical management of symptomatic, untreated patients with CLL.
Introduction
Chronic lymphocytic leukemia (CLL) is the most prevalent adult leukemia.1 At diagnosis, most patients are asymptomatic, and observation is the standard management approach.2 During watch and wait, patients with CLL have immune dysfunction and a high predisposition for infections and second primary cancers.3,4 Certain genomic features predict earlier progression, including unmutated immunoglobulin heavy-chain variable gene (IGHV) status, interphase cytogenetic abnormalities [del(17p), del(11q)], mutated tumor protein 53 (TP53), complex karyotype, and increased β2-microglobulin.5-9
Until recently, chemoimmunotherapy with fludarabine, cyclophosphamide, and rituximab, bendamustine and rituximab, or chlorambucil and obinutuzumab was the standard of care for treatment-naive patients who were physically fit (fludarabine, cyclophosphamide, and rituximab) or had coexisting conditions (chlorambucil plus obinutuzumab or bendamustine and rituximab).10-12 For patients with del(17p) or mutated TP53, however, chemoimmunotherapy regimens are generally not effective.13
Bruton tyrosine kinase (BTK) inhibitors significantly changed the treatment of CLL. Clinical studies of ibrutinib demonstrated efficacy in patients with relapsed CLL, including those with del(17p),14,15 and previously untreated patients with CLL.16 However, ibrutinib is associated with distinct adverse events (AEs), including diarrhea, rash, arthralgias, bleeding, hypertension, and atrial fibrillation, as well as treatment discontinuation owing to AEs in ∼8% to 19% of patients.14-17 These AEs are hypothesized to occur because of off-target effects of ibrutinib on alternative kinases, including epidermal growth factor receptor (EGFR), tyrosine kinase expressed in hepatocellular carcinoma (Tec), and interleukin-2–inducible T-cell kinase (ITK).18,19
Acalabrutinib (ACP-196) is a selective, next-generation covalent BTK inhibitor with less inhibition than of other kinases, such as EGFR, erythroblastosis oncogene B (ERBB2), Tec protein-tyrosine kinase, and ITK as compared with that seen with ibrutinib.19,20 Acalabrutinib was approved for treatment of patients with treatment-naive and relapsed/refractory CLL/small lymphocytic lymphoma (SLL) based on 2 phase 3 studies, ELEVATE-TN and ASCEND.21,22 The long-term efficacy and safety of acalabrutinib were demonstrated in the relapsed/refractory CLL cohort of the first phase 1/2 study of acalabrutinib (ACE-CL-001; #NCT02029443).20,23 Herein, we report the safety, efficacy, and prolonged follow-up from the initial ACE-CL-001 study in the treatment-naive CLL cohort.
Methods
The phase 1/2 multicenter study was designed to determine the recommended dose, safety, efficacy, pharmacokinetics, and pharmacodynamics of acalabrutinib in patients with symptomatic untreated CLL. All patients provided written informed consent. An institutional review board approved the protocol at each site. The study was registered at the clinical trials registry of the National Institutes of Health (#NCT02029443) and conducted according to the principles of the Declaration of Helsinki and International Conference on Harmonization Guidelines for Good Clinical Practice.
Patients
Eligibility included a diagnosis of previously untreated CLL/SLL requiring treatment per the 2008 International Workshop on Chronic Lymphocytic Leukemia guidelines24 ; adequate performance status (Eastern Cooperative Oncology Group performance status ≤2) and organ function; and absence of active infection. No restrictions for cytopenia were applied if CLL bone marrow involvement was present. Patients must have either declined or had comorbidities that precluded treatment with chemoimmunotherapy, as assessed by the investigator. Exclusion criteria included prior malignancy (except for adequately treated basal cell, squamous cell skin cancer, or in situ cervical cancer, or other prior malignancies for which the patient has been disease free for ≥2 years), need for warfarin or proton pump inhib‐itor therapy, active gastrointestinal inflammation or malabsorption, inadequate renal function (creatinine clearance <30 mL/min), and significant cardiovascular disease, such as uncontrolled or symptomatic arrhythmias, congestive heart failure, or myocardial infarction within 6 months of screening, and any New York Heart Association class III/IV cardiac disease. Patients must have had a left ventricular ejection fraction of >40% for inclusion (cardiac assessments conducted at screening included electrocardiogram and echocardiogram testing). Patients with prior or concurrent atrial fibrillation were eligible.
Evaluation and treatment
All patients had baseline assessment for interphase cytogenetics, IGHV mutation analysis, β2-microglobulin, and B symptoms. Complex karyotype was defined as ≥3 clonal chromosome abnormalities based on stimulated karyotype. Patients were successively enrolled to cohorts administering oral acalabrutinib at 200 mg once daily or 100 mg twice daily as part of the phase 2 portion of the study. An amendment converted all patients to the 100-mg twice-daily dose of acalabrutinib. The data cutoff for analysis reported herein is 1 August 2019.
Disease evaluation
Patients were evaluated at screening, weekly for the first month, biweekly for the second month, monthly for 4 months, and every 3 months thereafter with medical history, physical examinations, and laboratory studies for signs of toxicity. T cell, natural killer cell, and monocyte numbers, as well as serum immunoglobulins, were measured at baseline and before cycles 3, 10, and 16, then every 6 cycles until cycle 36, and then every 12 cycles thereafter.
Baseline assessments included central analysis of genomic aberrations with fluorescence in situ hybridization (FISH). In addition, IGHV and TP53 mutations were analyzed by DNA sequencing. A central laboratory tested peripheral blood samples obtained at baseline for all patients. FISH probes for cytogenetic profiling were used to test for abnormalities in chromosomes 13q, 12, 11q, and 17p (Vysis CLL FISH Probe Kit; Abbott Molecular, Chicago, IL). Where FISH data were not available from the central laboratory, results from local laboratories (n = 25) were used. IGHV mutation analysis was performed with standard Sanger sequencing. For TP53 mutation analyses, both Sanger and next-generation sequencing methods were used, as the central laboratory changed during the study; assay sensitivities were 20% and 5%, respectively, and average coverage for all amplicons was >1000 reads.
Computed tomographic (CT) scan (with contrast unless contraindicated) of the chest, abdomen, and pelvis was required during screening. CT scans could be performed as part of radiographic tumor assessment at the end of cycles 2, 4, and 6, then every 6 cycles until cycle 36, and then every 12 cycles thereafter. A CT scan was to be performed to confirm any case of suspected disease progression outside of these scheduled assessments. Changes in the size of lymph nodes, liver, and spleen were evaluated as part of the tumor response assessments at these time points.
AEs were graded according to the Common Terminology Criteria for Adverse Events, version 4.03. Response assessments, including radiologic examinations, were performed at the end of cycles 2, 4, and 6, then every 6 cycles until cycle 36, and then every 12 cycles thereafter. Bone marrow biopsy was done in all patients at 12 months or when all other criteria were met for complete response (CR). Response was evaluated based on the 2008 International Workshop on Chronic Lymphocytic Leukemia criteria with incorporation of the clarification for treatment-related lymphocytosis.24,25 Isolated elevation of treatment-related lymphocytosis following initiation of acalabrutinib was not considered progressive disease (PD) unless it was associated with a patient becoming symptomatic. A partial response (PR) in the setting of persistent lymphocytosis was characterized as a PR with lymphocytosis (PR-L).
Pharmacokinetics and pharmacodynamics
Pharmacokinetic analyses were performed using a validated assay20 during cycle 1. BTK occupancy was measured in peripheral blood mononuclear cells (PBMCs) using a biotin-tagged analog probe at baseline, 4 hours postdose on days 1 and 8, and predose on days 1, 8, and 28.
Sample collection and PBMC isolation
Blood was drawn in heparin-coated vacutainer tubes just prior to dosing on days 1, 2, 8, and 28 of cycle 1 and 4 hours after dosing on days 1, 8, and 28 of cycle 1. Samples were shipped at ambient temperature overnight to Acerta Pharma analytical laboratories and immediately purified to obtain PBMCs using Ficoll Paque Plus density separation method (product insert instructions 71-7167-00 AG; GE Healthcare Biosciences AB; Uppsala, Sweden), followed by cryopreservation in liquid nitrogen.
BTK target occupancy
Detailed procedures are outlined in the supplemental Methods, available on the Blood Web site. In brief, samples from all time points from each patient were analyzed together with a control PBMC sample from a healthy volunteer. Occupancy was tested using an enzyme-linked immunosorbent assay–based method in PBMCs.
Statistical analyses
Safety and efficacy analyses included all enrolled patients who received ≥1 dose of study drug. Descriptive statistics are reported. Wilcoxon signed-rank test was used to assess changes from baseline in immunoglobulin levels and immune cell counts. Only patients with values at baseline and at each follow-up visit were included; analyses were not adjusted for multiplicity. P values <.05 were regarded as descriptive and not definitive because of the high number of exploratory tests performed. Progression-free survival (PFS) was defined as the time from the first dose to documentation of PD or death; duration of response (DOR) was defined as the time from the date of first response (PR-L or better) to documentation of PD or death (any cause). PFS and DOR values were estimated using Kaplan-Meier methodology26 ; PFS and DOR data were censored at the last clinical assessment for patients who discontinued treatment without disease progression or death. Event-free survival (EFS) was defined as time from the first dose to documented PD, death, treatment discontinuation owing to an AE, or start of new anticancer therapy.
Results
Patients
A total of 99 patients were enrolled to the treatment-naive study cohort between 15 August 2014 and 10 December 2015 and received ≥1 dose of acalabrutinib. The median patient age was 64 (range, 33-85) years; 67% were men, and 47% had high-risk disease as defined by Rai criteria27 (Table 1). Bulky lymph nodes ≥5 cm were noted in 46% of patients, and β2-microglobulin was >3.5 mg/mL in 77% of patients. Genomic features at time of study enrollment included 62% with unmutated IGHV (defined using a cutoff of 2% variance from germline sequence), 21% with del(11q), 10% with del(17p), 14% with mutated TP53, 18% with any TP53 aberration, and 18% with complex karyotype. The 37 patients who initially received acalabrutinib 200 mg once daily were treated for a median 13.7 months (range, 2.5-33.6 months) before crossing over to 100-mg twice-daily dosing (median, 41.5 months; range, 5.6-51.8 months). Because the duration of treatment on 200 mg once daily was brief compared with the duration on 100 mg twice daily, all analyses were conducted in the overall dosing population.
Baseline patient characteristics
| Characteristic | N = 99 |
|---|---|
| Median age, y (range) | 64 (33-85) |
| ≥65 y, n (%) | 45 (46) |
| Male sex, n (%) | 66 (67) |
| Histology, n (%) | |
| CLL | 98 (99) |
| SLL | 1 (1) |
| Median time from initial CLL diagnosis to first dose, y (range) | 3.4 (0.1-16.5) |
| Baseline Rai stage, n (%) | |
| I to II | 43 (43) |
| III to IV | 47 (47) |
| Unknown | 9 (9) |
| ECOG PS score, n (%) | |
| 0 | 34 (34) |
| 1 | 65 (66) |
| Patients with cytopenia(s), n (%) | 52 (53) |
| Absolute neutrophil count ≤1.5 × 109/L | 10 (10) |
| Hemoglobin ≤11 g/dL | 38 (38) |
| Platelets ≤100 × 109/L | 28 (28) |
| Presence of B symptoms, n (%) | 21 (21) |
| β2-microglobulin >3.5 mg/L, n/N (%) | 72/93 (77) |
| Bulky disease, n/N (%) | |
| ≥5 cm | 46/99 (46) |
| ≥10 cm | 6/99 (6) |
| Genomic status, n/n (%) | |
| del(11q)* | 19/91 (21) |
| del(17p)† | 9/91 (10) |
| Complex karyotype | 12/66 (18) |
| Unmutated IGHV | 57/92 (62) |
| Mutated TP53 | 9/65 (14) |
| TP53 aberration‡ | 12/68 (18) |
| Characteristic | N = 99 |
|---|---|
| Median age, y (range) | 64 (33-85) |
| ≥65 y, n (%) | 45 (46) |
| Male sex, n (%) | 66 (67) |
| Histology, n (%) | |
| CLL | 98 (99) |
| SLL | 1 (1) |
| Median time from initial CLL diagnosis to first dose, y (range) | 3.4 (0.1-16.5) |
| Baseline Rai stage, n (%) | |
| I to II | 43 (43) |
| III to IV | 47 (47) |
| Unknown | 9 (9) |
| ECOG PS score, n (%) | |
| 0 | 34 (34) |
| 1 | 65 (66) |
| Patients with cytopenia(s), n (%) | 52 (53) |
| Absolute neutrophil count ≤1.5 × 109/L | 10 (10) |
| Hemoglobin ≤11 g/dL | 38 (38) |
| Platelets ≤100 × 109/L | 28 (28) |
| Presence of B symptoms, n (%) | 21 (21) |
| β2-microglobulin >3.5 mg/L, n/N (%) | 72/93 (77) |
| Bulky disease, n/N (%) | |
| ≥5 cm | 46/99 (46) |
| ≥10 cm | 6/99 (6) |
| Genomic status, n/n (%) | |
| del(11q)* | 19/91 (21) |
| del(17p)† | 9/91 (10) |
| Complex karyotype | 12/66 (18) |
| Unmutated IGHV | 57/92 (62) |
| Mutated TP53 | 9/65 (14) |
| TP53 aberration‡ | 12/68 (18) |
Positive for del(11q) without or missing del(17p) [1 patient had missing del(17p) status].
Positive for del(17p) with or without del(11q).
Includes TP53 mutations or del(17p).
ECOG PS, Eastern Cooperative Oncology Group performance status; SLL, small lymphocytic leukemia.
Safety
Acalabrutinib was administered for a median of 52 months (range, 0.2-60); the overall median time on treatment was 53 months (range, 1-59) (56 months for 200 mg once daily [n = 37] and 50 months for 100 mg twice daily [n = 62]). At the time of this report, 85 patients (86%) remained on therapy. Of the 14 patients who discontinued therapy, the most common reason was AEs (n = 6; second primary cancers [n = 4] and infection [n = 2]). Treatment discontinuations because of second primary cancers included angiosarcoma, glioblastoma multiforme, small-cell lung cancer, and prostate cancer (n = 1 each); those due to infections were sepsis (grade 4; n = 1) and urinary tract infection (grade 3; n = 1). Other reasons included disease progression (n = 3), withdrawal of consent (n = 2), pregnancy (n = 1), and start of other anticancer therapy (n = 1). Two patients (2%) died while on study, one because of multiple organ dysfunction (in the setting of pneumonia in the same patient who experienced grade 4 sepsis) and one because of cardiac failure during an ongoing lung infection and atrial fibrillation.
The most common AEs (≥20% all grades) included diarrhea, headache, upper respiratory tract infection, contusion, arthralgia, weight increase, nausea, cough, vomiting, hypertension, and sinusitis (Table 2); most were grade ≤2. Median weight was 82.7 kg (n = 98; range, 45.9-154.5) at baseline and 87.6 kg (n = 85; range, 45.9-148.7) by cycle 48 (supplemental Figure 1). AE incidence diminished over time on study (Figure 1). Serious AEs were reported in 38 patients (38%); the most common serious AEs (≥2 patients) were pneumonia (n = 4), sepsis (n = 3), and influenza, pancreatitis, prostate cancer, pyrexia, sinusitis, and urinary tract infection (n = 2 each).
AEs occurring in ≥15% of patients
| All treated patients (N = 99) | ||||
|---|---|---|---|---|
| AE, n (%)* | All grades | Grade 1 | Grade 2 | Grade 3 |
| Diarrhea | 51 (51) | 37 (37) | 9 (9) | 5 (5) |
| Headache | 45 (45) | 34 (34) | 6 (6) | 5 (5) |
| Upper respiratory tract infection | 44 (44) | 5 (5) | 38 (38) | 1 (1) |
| Arthralgia | 42 (42) | 29 (29) | 12 (12) | 1 (1) |
| Contusion | 42 (42) | 38 (38) | 4 (4) | 0 |
| Weight increased | 32 (32) | 15 (15) | 14 (14) | 3 (3) |
| Nausea | 31 (31) | 23 (23) | 4 (4) | 4 (4) |
| Cough | 30 (30) | 22 (22) | 8 (8) | 0 |
| Vomiting | 22 (22) | 17 (17) | 3 (3) | 2 (2) |
| Hypertension | 21 (21) | 4 (4) | 7 (7) | 10 (10) |
| Sinusitis | 21 (21) | 2 (2) | 17 (17) | 2 (2) |
| Fall | 19 (19) | 13 (13) | 5 (5) | 1 (1) |
| Fatigue | 19 (19) | 15 (15) | 3 (3) | 1 (1) |
| Back pain | 18 (18) | 9 (9) | 9 (9) | 0 |
| Nasal congestion | 18 (18) | 12 (12) | 6 (6) | 0 |
| Petechiae | 18 (18) | 17 (17) | 1 (1) | 0 |
| Rash | 17 (17) | 14 (14) | 3 (3) | 0 |
| Constipation | 16 (16) | 14 (14) | 2 (2) | 0 |
| Ecchymosis | 16 (16) | 15 (15) | 1 (1) | 0 |
| Gastroesophageal reflux disease | 16 (16) | 10 (10) | 6 (6) | 0 |
| Oropharyngeal pain | 16 (16) | 12 (12) | 4 (4) | 0 |
| Night sweats | 15 (15) | 11 (11) | 4 (4) | 0 |
| Peripheral edema | 15 (15) | 12 (12) | 3 (3) | 0 |
| All treated patients (N = 99) | ||||
|---|---|---|---|---|
| AE, n (%)* | All grades | Grade 1 | Grade 2 | Grade 3 |
| Diarrhea | 51 (51) | 37 (37) | 9 (9) | 5 (5) |
| Headache | 45 (45) | 34 (34) | 6 (6) | 5 (5) |
| Upper respiratory tract infection | 44 (44) | 5 (5) | 38 (38) | 1 (1) |
| Arthralgia | 42 (42) | 29 (29) | 12 (12) | 1 (1) |
| Contusion | 42 (42) | 38 (38) | 4 (4) | 0 |
| Weight increased | 32 (32) | 15 (15) | 14 (14) | 3 (3) |
| Nausea | 31 (31) | 23 (23) | 4 (4) | 4 (4) |
| Cough | 30 (30) | 22 (22) | 8 (8) | 0 |
| Vomiting | 22 (22) | 17 (17) | 3 (3) | 2 (2) |
| Hypertension | 21 (21) | 4 (4) | 7 (7) | 10 (10) |
| Sinusitis | 21 (21) | 2 (2) | 17 (17) | 2 (2) |
| Fall | 19 (19) | 13 (13) | 5 (5) | 1 (1) |
| Fatigue | 19 (19) | 15 (15) | 3 (3) | 1 (1) |
| Back pain | 18 (18) | 9 (9) | 9 (9) | 0 |
| Nasal congestion | 18 (18) | 12 (12) | 6 (6) | 0 |
| Petechiae | 18 (18) | 17 (17) | 1 (1) | 0 |
| Rash | 17 (17) | 14 (14) | 3 (3) | 0 |
| Constipation | 16 (16) | 14 (14) | 2 (2) | 0 |
| Ecchymosis | 16 (16) | 15 (15) | 1 (1) | 0 |
| Gastroesophageal reflux disease | 16 (16) | 10 (10) | 6 (6) | 0 |
| Oropharyngeal pain | 16 (16) | 12 (12) | 4 (4) | 0 |
| Night sweats | 15 (15) | 11 (11) | 4 (4) | 0 |
| Peripheral edema | 15 (15) | 12 (12) | 3 (3) | 0 |
No grade 4 or 5 events reported.
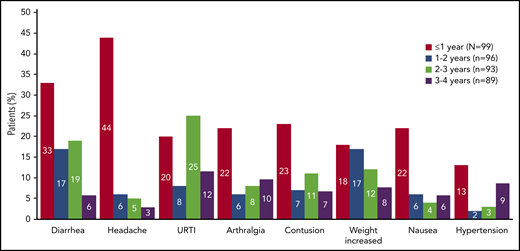
Incidence of select TEAEs by yearly intervals. The incidence of TEAEs selected by frequency (≥15% at ≤1 year) and clinical interest. TEAE, treatment-emergent adverse event; URTI, upper respiratory tract infection.
Incidence of select TEAEs by yearly intervals. The incidence of TEAEs selected by frequency (≥15% at ≤1 year) and clinical interest. TEAE, treatment-emergent adverse event; URTI, upper respiratory tract infection.
AEs of clinical interest, including atrial fibrillation, hypertension, bleeding, infection events, and headache, were assessed to further explore the safety profile of acalabrutinib (Table 3).
ECI
| ECI* | All treated patients (N = 99), n (%) | |
|---|---|---|
| All grades | Grade ≥3 | |
| Cardiac events | 20 (20)† | 4 (4) |
| Atrial fibrillation | 5 (5)‡ | 2 (2) |
| Ventricular tachyarrhythmias | 0 | 0 |
| Anemia | 8 (8) | 2 (2) |
| Leukopenia | 9 (9) | 9 (9) |
| Neutropenia | 9 (9) | 9 (9) |
| Other leukopenia | 1 (1) | 1 (1) |
| Thrombocytopenia | 3 (3) | 1 (1) |
| Hemorrhage§ | 65 (66) | 3 (3) |
| Major hemorrhage‖ | 4 (4) | 3 (3) |
| Hepatotoxicity | 4 (4) | 2 (2) |
| Hypertension | 22 (22) | 11 (11) |
| Infections¶,# | 83 (84) | 15 (15) |
| Interstitial lung disease/pneumonitis | 1 (1) | 0 |
| Second primary malignancies | 26 (26) | 5 (5) |
| Second primary malignancies, excluding nonmelanoma skin | 11 (11) | 5 (5) |
| Tumor lysis syndrome | 0 | 0 |
| ECI* | All treated patients (N = 99), n (%) | |
|---|---|---|
| All grades | Grade ≥3 | |
| Cardiac events | 20 (20)† | 4 (4) |
| Atrial fibrillation | 5 (5)‡ | 2 (2) |
| Ventricular tachyarrhythmias | 0 | 0 |
| Anemia | 8 (8) | 2 (2) |
| Leukopenia | 9 (9) | 9 (9) |
| Neutropenia | 9 (9) | 9 (9) |
| Other leukopenia | 1 (1) | 1 (1) |
| Thrombocytopenia | 3 (3) | 1 (1) |
| Hemorrhage§ | 65 (66) | 3 (3) |
| Major hemorrhage‖ | 4 (4) | 3 (3) |
| Hepatotoxicity | 4 (4) | 2 (2) |
| Hypertension | 22 (22) | 11 (11) |
| Infections¶,# | 83 (84) | 15 (15) |
| Interstitial lung disease/pneumonitis | 1 (1) | 0 |
| Second primary malignancies | 26 (26) | 5 (5) |
| Second primary malignancies, excluding nonmelanoma skin | 11 (11) | 5 (5) |
| Tumor lysis syndrome | 0 | 0 |
ECIs, events of clinical interest; UTI, urinary tract infection.
ECIs were based on combined AE terms for infections, bleeding events, hypertension, and second primary malignancies, excluding nonmelanoma skin and on a single AE term for atrial fibrillation.
Other cardiac events occurring in >1 patient included tachycardia (n = 8; all grade 1), sinus tachycardia (n = 3; all grade 1), palpitations (n = 3 [grade 1, n = 2; grade 2, n = 1]), and angina pectoris (n = 2; both grade 2).
Age range: 68 to 79 y.
Most common bleeding events (all grade, ≥15%) were contusion (42%), petechiae (18%), and ecchymosis (16%).
Includes 1 grade 1 AE (retinal hemorrhage) and 3 grade 3 AEs (hematuria, traumatic intracranial hemorrhage, and upper gastrointestinal hemorrhage).
Most common infections (all grade, ≥15%) were upper respiratory tract infections (44%) and sinusitis (21%); among patients with opportunistic infections, grade 2 fungal infection, grade 2 coccidioidomycosis, and grade 2 herpes zoster were reported in 1 patient each, and 1 patient experienced both grade 3 perineal infection fungal and grade 3 herpes zoster.
Acalabrutinib dosing was delayed in 10 patients because of infection AEs; 2 patients discontinued acalabrutinib because of grade 3 UTI and grade 4 sepsis.
Eight atrial fibrillation events (grade 2, n = 5; grade 3, n = 3) were reported in 5 patients (5% all grades). All 5 patients had a medical history of hypertension and de novo disease and received treatment aimed at either rhythm and/or rate control (amiodarone or metoprolol); two of these patients also received long-term anticoagulation treatment (rivaroxaban or enoxaparin). The 8 atrial fibrillation events occurred on days 8, 724, 736, 823, 838, 1213, 1214, and 1241 (the latter 4 events occurred in 1 patient who died because of cardiac failure). The patient with grade 3 atrial fibrillation on study day 8 experienced acute myocardial infarction 6 days after this event and underwent coronary artery bypass grafting 2 days later; acalabrutinib was withheld for 56 days. The overall atrial fibrillation incidence rate was 2.0 per 100 person-years. No patient discontinued acalabrutinib because of atrial fibrillation.
Hypertension (all grades) occurred in 22 patients (22%). Of these patients, 10 had preexisting disease. Fourteen of the 22 patients received no antihypertensive medication while on study treatment. Most patients had low-grade hypertension; 11% had grade ≥3 events. The most common drugs started on study to treat hypertension were calcium channel blockers (amlodipine, verapamil) in 4 patients, beta-blockers (metoprolol) in 3 patients, angiotensin receptor antagonists (irbesartan, telmisartan, losartan) in 3 patients, and angiotensin-converting enzyme inhibitors in combination with potassium-sparing diuretics in 2 patients. Median time to first onset of hypertension was 169 days (range, 3-1681). No patient discontinued acalabrutinib because of hypertension. During the time of observation (as of data cutoff), no patient suffered sudden unexplained death, including sudden cardiac death.
Bleeding events (all grades) occurred in 65 patients (66%); the most common bleeding events (≥15% all grades) were contusion (42%), petechiae (18%), and ecchymosis (16%). Grade ≥3 hemorrhage occurred in 3 patients (3%) (hematuria, traumatic intracranial hemorrhage, and upper gastrointestinal hemorrhage). Acalabrutinib was withheld for 2 patients (concurrent grade 2 contusion and grade 2 scrotal ecchymosis [5 days] and grade 3 upper gastrointestinal hemorrhage [13 days]). No patient discontinued acalabrutinib because of bleeding events.
Infections (all grades) occurred in 83 patients (84%); the most common infections (≥15% all grades) were upper respiratory tract infections (44%) and sinusitis (21%). Fifteen patients (15%) had grade 3 or 4 infection; among them, influenza virus, herpes zoster, and fungal infection were reported in 1 patient each. Acalabrutinib was withheld in 15 patients during these infections; 1 patient each discontinued acalabrutinib because of grade 3 urinary tract infection and grade 4 sepsis.
Headache AEs (all grades) were reported in 45 patients (45%); most were grade 1 (n = 34/45). Headache AEs were considered acalabrutinib related by the investigator in 34 patients (grade 1, n = 27; grade 2, n = 4; grade 3, n = 3). Median time to first onset was 7 days (range, 1-775). Multiple headaches (≥2 events) occurred in 17 patients (17%). Median duration of headache events (n = 73) was 43 days (range, 1- 1508). No patient discontinued because of headache.
Immunologic studies
Assessment of immune effector cells showed statistically significant decreases in CD4 (T helper), CD8 (T suppressor), and natural killer cells, with no change from baseline in monocyte levels; values remained within the normal ranges throughout treatment (supplemental Figure 2). Immunoglobulin levels were stable over time with the exception of a statistically significant (P < .05) rise in immunoglobulin A level that remained within the normal range (supplemental Figure 3).
Response to acalabrutinib
The overall response rate (ORR), including CR, PR, and PR-L, was 97% (Figure 2A). Best response was CR in 7% of patients and PR in 90% (no patient had best response of PR-L). The median time to first response was 3.7 months (range, 2-22); the median time to CR was 33 months (range, 22-55). The ORR for each high-risk subgroup (unmutated IGHV, del(17p), mutated TP53, complex karyotype) was 100%. Improvement in cytopenias to normal levels occurred in 97% (n = 37/38) of patients who had anemia prior to starting treatment (median time to recovery to normal levels: 85 days), 100% (n = 10 of 10) of those with neutropenia (median: 9 days), and 96% (n = 27 of 28) of those with thrombocytopenia (median: 22 days). Greater than 50% reduction in lymphadenopathy was noted in all patients except one who had a best response of stable disease (Figure 2B).
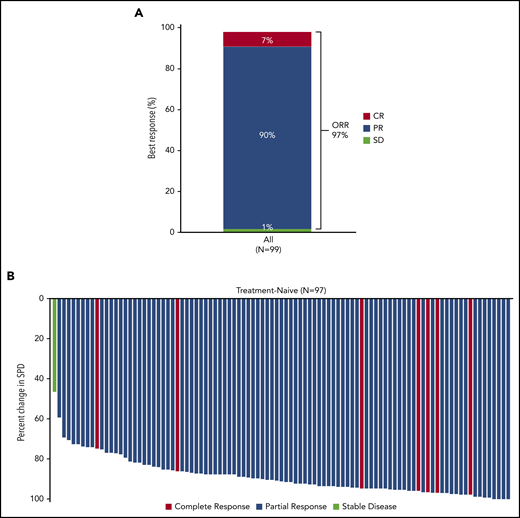
Response to acalabrutinib and best change in tumor size. (A) ORR was assessed in all patients who received ≥1 dose of acalabrutinib. (B) Waterfall plot showing the maximum percent change from baseline in the sum of product diameter (SPD) by investigator assessment for all treated patients with tumor assessments (N = 97). Two patients without postbaseline overall response assessment were excluded. SD, stable disease.
Response to acalabrutinib and best change in tumor size. (A) ORR was assessed in all patients who received ≥1 dose of acalabrutinib. (B) Waterfall plot showing the maximum percent change from baseline in the sum of product diameter (SPD) by investigator assessment for all treated patients with tumor assessments (N = 97). Two patients without postbaseline overall response assessment were excluded. SD, stable disease.
Response duration, PFS, and EFS
The median DOR has not been reached; the estimated 48-month DOR was 97% (95% confidence interval [CI], 90-99). Median PFS has not been reached (Figure 3A); the estimated 48-month PFS rate is 96% (95% CI, 89-98). Median EFS also was not reached (Figure 3B); the estimated 48-month EFS rate was 90% (95% CI, 82-94). Median PFS and EFS in patients with del(17p) and/or mutated TP53 (n = 12) and complex karyotype (n = 12) were not reached (Figure 4). The estimated 48-month PFS and EFS rates were 82% and 75%, respectively, in patients with del(17p) and/or mutated TP53 and 91% and 83%, respectively, in patients with complex karyotype. Three patients discontinued acalabrutinib due to PD; the time from first acalabrutinib dose to disease progression was 25 months [patient with del(11q)], 32 months [patient with del(17p), unmutated IGHV, complex karyotype, and Richter transformation], and 40 months [patient with del(17p), unmutated IGHV, and mutated TP53]. Patients were not followed on study after progression or treatment discontinuation.
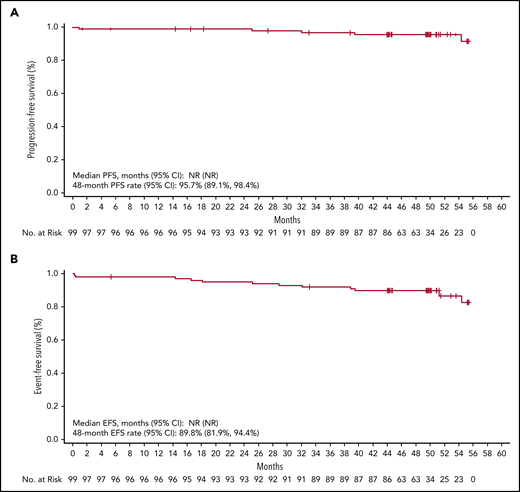
PFS and EFS. Shown are data for PFS (A) and EFS (B) with Kaplan-Meier estimates in all treated patients (N = 99). NR, not reached.
PFS and EFS. Shown are data for PFS (A) and EFS (B) with Kaplan-Meier estimates in all treated patients (N = 99). NR, not reached.
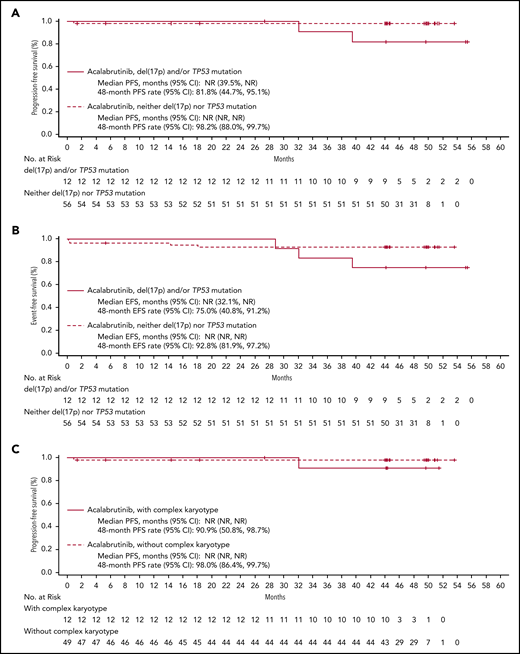
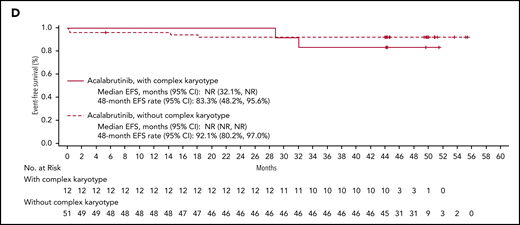
PFS and EFS in patients with high-risk features. Shown are Kaplan-Meier curves for PFS and EFS by del(17p) and TP53 mutation status (A and B, respectively) and by complex karyotype (C and D, respectively).
PFS and EFS in patients with high-risk features. Shown are Kaplan-Meier curves for PFS and EFS by del(17p) and TP53 mutation status (A and B, respectively) and by complex karyotype (C and D, respectively).
Pharmacodynamic studies
BTK occupancy in circulating blood mononuclear cells was assessed in 35 patients receiving once-daily dosing and 52 patients receiving twice-daily dosing. Median BTK occupancy of 97% to 99% was observed throughout twice-daily dosing at steady state on days 8 and 28 at trough, which was significantly better than that with 200 mg once-daily dosing (at trough day 8 [P = .0034] and day 28 [P = .0003]; 100 mg twice daily vs 200 mg once daily; Figure 5).
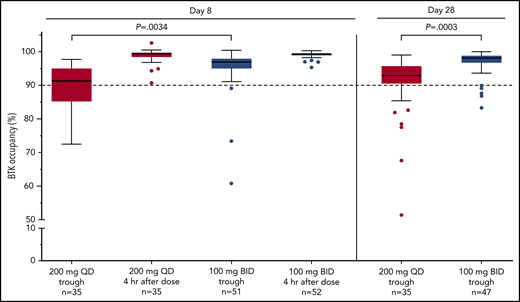
BTK occupancy. The level of drug binding to BTK over time in PBMCs. BTK target occupancy ELISA was performed on PBMC lysates using biotin-tagged probes. The figure shows data from the assessment at day 8 prior to the subsequent dose (steady-state trough) and day 28. “Pre” refers to time point preceding the morning dose. “Post” refers to 4 hours after the dose. The horizontal lines indicate the median value. Statistical significance was determined using an unpaired, parametric, 2-tailed Student t test (descriptive P values were reported). BID, twice daily; ELISA, enzyme-linked immunoassay; QD, once daily.
BTK occupancy. The level of drug binding to BTK over time in PBMCs. BTK target occupancy ELISA was performed on PBMC lysates using biotin-tagged probes. The figure shows data from the assessment at day 8 prior to the subsequent dose (steady-state trough) and day 28. “Pre” refers to time point preceding the morning dose. “Post” refers to 4 hours after the dose. The horizontal lines indicate the median value. Statistical significance was determined using an unpaired, parametric, 2-tailed Student t test (descriptive P values were reported). BID, twice daily; ELISA, enzyme-linked immunoassay; QD, once daily.
Discussion
Herein, we provide data for long-term follow-up in 99 patients with symptomatic, previously untreated CLL who were treated with acalabrutinib. Enrolled patients had Rai stage and genomic features representative of other trials in previously untreated CLL.16,28 Acalabrutinib demonstrated a 97% ORR, including 7% of patients with CR; the estimated PFS and EFS at 4 years was 96% and 90%, respectively. Durable responses were also noted across all prognostic groups, including those with del(17p) and mutated TP53. Acalabrutinib was associated with a favorable safety profile, with few treatment discontinuations due to AEs (6%). At a median follow-up of 53 months, 86% of patients remain on acalabrutinib. Collectively, these data provide robust evidence that long-term acalabrutinib monotherapy is efficacious in symptomatic, previously untreated CLL. This work provides long-term data on the safety and efficacy of acalabrutinib in treatment-naive CLL.
Biochemical and cellular studies of acalabrutinib demonstrate favorable selectivity for irreversible inhibition of BTK with essentially no irreversible inhibition of erythroblastosis oncogene B, EGFR1, and ITK, and a greater therapeutic window between BTK and Tec protein-tyrosine kinase compared with ibrutinib.20 In the present study, acalabrutinib treatment was associated with a reduction in CD4 and CD8 cells that remained within the normal ranges, whereas an increase in these cell types has been reported with ibrutinib.29 The clinical relevance of the differential impact on T cells is currently unknown, but it is possible that the observed differences may be related to differences in target specificity between the 2 agents.29 The increased selectivity and short half-life of acalabrutinib allow treatment of CLL with greater specificity for BTK inhibition.30,31 The pharmacodynamic data, including data from the relapsed/refractory cohort,20,32 demonstrated increased BTK occupancy with twice-daily vs once-daily dosing, leading to the change in schedule to twice-daily dosing for all patients enrolled in the trial. Recent data from lymph node biopsy assessment demonstrate improved reduction of B-cell receptor signaling in this compartment with acalabrutinib twice-daily vs once-daily dosing, supporting this schedule change.32
AEs observed in this trial were generally mild; only a small subset of patients were withdrawn because of drug toxicity. AEs generally resolved over time. Common to acalabrutinib was headache (grade 1 in most cases). Weight gain was noted in approximately one-third of patients, which has previously been reported with ibrutinib.33 The mechanism behind the weight gain is currently unknown, but it may be a class effect of BTK inhibitors and/or because of the reversal of CLL-induced cachexia through successful treatment with targeted, noncytotoxic therapy33 ; however, additional research is needed. AEs of special interest identified based on initial ibrutinib studies were generally low grade. In some cases, AEs of special interest occurred less frequently than has been reported for ibrutinib; for example, compared with an incidence of 5% in our analysis, atrial fibrillation (all grades) was reported in 11% of patients in an integrated analysis of single-agent ibrutinib (treatment duration up to 43 months) from 3 pivotal studies in CLL.34 For the long-term RESONATE-2 study, which evaluated ibrutinib monotherapy in treatment-naive patients, all-grade atrial fibrillation was 16% (grade 3, 5%) after 60 months of follow-up.35 The incidence of hypertension reported for acalabrutinib in this study (grade ≥3 incidence of 10%) could be an underestimation of the potential incidence with longer follow-up (grade ≥3 incidence was 32% at 5 years with ibrutinib15 ); however, long-term follow-up of RESONATE-2 showed an all-grade rate of hypertension of 26% and grade 3 hypertension in only 9% of patients.35 A randomized phase 3 trial comparing ibrutinib with acalabrutinib in previously treated, high-risk patients with CLL (#NCT02477696) completed enrollment and will compare the frequency and severity of AEs.
This trial reports a high response rate and durable remissions with single-agent acalabrutinib. A phase 3 study comparing acalabrutinib, acalabrutinib and obinutuzumab, and chlorambucil and obinutuzumab in patients with treatment-naive CLL (ELEVATE-TN) reported improved outcomes with both acalabrutinib-containing arms compared with those seen with chlorambucil and obinutuzumab.22 Although the ELEVATE-TN study was not designed to examine comparative outcomes between acalabrutinib alone and combined acalabrutinib plus obinutuzumab, it is notable that in a post hoc analysis, a PFS benefit was suggested with the addition of obinutuzumab to acalabrutinib (hazard ratio, 0.49; 95% CI, 0.26-0.95).22 Whether the PFS benefit continues with longer follow-up and which patients are most likely to derive this benefit remain to be seen. The ORR reported for acalabrutinib in the current study (97%) was generally in line with the ORR for acalabrutinib monotherapy in ELEVATE-TN (86%; median treatment duration, 28.3 months).22 Similarly, a response rate of 92% was reported for ibrutinib monotherapy in the long-term RESONATE-2 study in treatment-naive patients with CLL (median treatment duration, 60 months).35 However, there are limitations in comparing the results of the current analysis, which was based on a phase 1/2 study conducted predominantly at academic centers in the United States, with those of larger phase 3, global studies that differ in patient populations and numbers, regional representation, and treatment duration.
In summary, this phase 1/2 trial of acalabrutinib in patients with previously untreated CLL demonstrates favorable safety, response, and durability of response among all prognostic groups. This trial provides suggestive evidence for improved long-term tolerability and low discontinuation rates with acalabrutinib monotherapy. The long durability of remission seen in this trial, along with most patients remaining on therapy, suggests that acalabrutinib represents an acceptable treatment option for patients with treatment-naive CLL and should be further evaluated in head-to-head trials with other standard-of-care therapies in this population.
Acerta Pharma, a member of the AstraZeneca Group, is committed to data transparency and will consider data sharing requests on a case-by-case basis. Any requests for deidentified patient data can be submitted to Acerta Pharma 3 months postpublication and ending 5 years following article publication with the intent to achieve aims of the original proposal. In addition, Acerta Pharma will provide the study protocol, statistical analysis plan, informed consent form as well as post results on clinicaltrials.gov, as required.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients who participated in this study and their families, the investigators, and coordinators at each of the clinical sites. Medical writing assistance, funded by Acerta Pharma, was provided by Tracy Diaz and Cindy Gobbel of Peloton Advantage, LLC, an OPEN Health company, under the direction of the authors.
This project was supported National Institutes of Health, National Cancer Institute grant R35 CA197734 (J.C. Byrd), The Connie Brown CLL Foundation, the Kevin Sullivan Foundation, the D. Warren Brown Foundation, and the Four Winds Foundation.
This study was funded by Acerta Pharma (South San Francisco, CA), a member of the AstraZeneca Group. Acerta Pharma provided the study drug. The clinical study was designed by A.H. and R.I. (Acerta Pharma) in collaboration with J.C. Byrd (The Ohio State University Comprehensive Cancer Center, Columbus, OH). Data collection and interpretation were performed by the authors, investigators, and study sponsor. The study sponsor reviewed the manuscript for accuracy.
Authorship
Contribution: J. C. Byrd, R.I., and W.G.W. provided substantial contributions to the design of the study; J. C. Byrd, J.A.W., R.R.F., P.M., S.O., J.R.B., D.M.S., J. C. Barrientos, S.D., P.H., J.M.P., A.H., R.I., P.P., M.H.W., N.J., and W.G.W. contributed to data collection and interpretation; M.H.W. performed statistical analyses; J. C. Byrd had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis; and all authors reviewed and provided important intellectual contributions to the manuscript and approved the final version for publication.
Conflict-of-interest disclosure: J. C. Byrd has received research funding from Janssen, Genentech, Pharmacyclics, and Acerta. J.A.W. served in a consulting or advisory role for AbbVie, Pharmacyclics, Janssen, AstraZeneca, ArQule; and has received research funding from Janssen, Karyopharm Therapeutics, MorphoSys, Verastem, Loxo, AbbVie. R.R.F. served in a consulting or advisory role for Pharmacyclics, Janssen Biotech, Genentech/Roche, Sunesis Pharmaceuticals, Loxo, TG Therapeutics, Verastem, Acerta Pharma, AstraZeneca, BeiGene, Incyte, OncoTracker, AbbVie; and has received fees for travel, accommodations, expenses from TG Therapeutics, Janssen Oncology; provided expert testimony for AbbVie, Janssen Oncology; maintains other relationships with Incyte, Janssen Biotech, AbbVie; has received honoraria from Janssen, Genentech/Roche; and has received research funding from Acerta Pharma, TG Therapeutics. P.M. served in a consulting or advisory role for AstraZeneca, Celgene, Janssen, Bayer, Kite Pharma, BeiGene, Celldex, Cellectar, I-Mab, MorphoSys, Regeneron, Sandoz, TeneoBio, Verastem, Karyopharm Therapeutics; has received fees for travel, accommodations, expenses from Janssen, MorphoSys; and has received research funding (the Institution) from Karyopharm Therapeutics. S.O. is employed by University of California Irvine; served in a consulting or advisory role for Amgen, Celgene, GlaxoSmithKline, Janssen Oncology, Aptose Biosciences, Vaniam Group, AbbVie/Genentech, Sunesis Pharmaceuticals, Alexion Pharmaceuticals, Astellas Pharma, Gilead Sciences, Pharmacyclics, TG Therapeutics, Pfizer, Verastem, Eisai, Juno Therapeutics, Vida Ventures; has received fees for travel, accommodations, expenses from Celgene, Janssen, Gilead Sciences, Regeneron, Janssen Oncology; has received honoraria from Celgene, Janssen, Pharmacyclics, Gilead Sciences, Pfizer, Amgen, Astellas Pharma, GlaxoSmithKline, Aptose Biosciences, Vaniam Group, AbbVie, Sunesis Pharmaceuticals, Alexion Pharmaceuticals, Loxo, Eisai, TG Therapeutics; has received research funding (the Institution) from Acerta Pharma, Regeneron, Gilead Sciences, Pfizer, TG Therapeutics, Pharmacyclics, Kite Pharma, Sunesis Pharmaceuticals. J.R.B. served in a consulting or advisory role for AbbVie, Acerta, AstraZeneca, BeiGene, Catapult, Dynamo, Eli Lilly, Genentech/Roche, Gilead, Juno/Celgene, Kite, Loxo, MEI Pharma, Nextcea, Novartis, Octapharma, Rigel, Pfizer, Pharmacyclics, Redx, Sun, Sunesis, TG Therapeutics, Verastem; has received honoraria from Janssen and Teva; has received research funding from Gilead, Loxo, Sun, Verastem; and has served on data safety monitoring committees for Morphosys, Invectys. D.M.S. served in a consulting or advisory role for Pharmacyclics/Janssen, Karyopharm, BeiGene, Innate; has received honoraria from Genentech; has received research funding from Acerta Pharma, Gilead Sciences, Karyopharm Therapeutics, Verastem, Juno Therapeutics. J. C. Barrientos served in a consulting or advisory role for Pharmacyclics, AbbVie, AstraZeneca, Gilead Sciences, Genentech, Bayer, Sandoz; has received fees for travel, accommodations, expenses from Janssen; has received honoraria from Janssen; has received research funding (the Institution) from Pharmacyclics, Oncternal Therapeutics, AstraZeneca. P.H. has received fees for travel, accommodations, expenses from Janssen, AbbVie; has received honoraria from Janssen, AbbVie, Roche; has received research funding (the Institution) from Janssen, Pharmacyclics, Roche, Gilead Sciences. J.M.P. served in a consulting or advisory role for Pharmacyclics, Gilead Sciences, AstraZeneca, Actinium Pharmaceuticals. A.H. and R.I. hold stock ownership/patents for Acerta Pharma and are former employees of Acerta at the time of the study. P.P. and M.H.W. are employed or hold stock ownership in Acerta Pharma/AstraZeneca. N.J. served in a consulting or advisory role for Pharmacyclics, Janssen, AbbVie, Genentech, AstraZeneca, Verastem, Adaptive Biotechnologies, Servier, Precision Biosciences, TG Therapeutics, BeiGene; has received research funding (the Institution) from Pharmacyclics, AbbVie, Genentech, AstraZeneca, Bristol Myers Squibb, Pfizer, ADC Therapeutics, Incyte, Servier, Cellectis, Verastem, Adaptive Biotechnologies, Precision Biosciences, Fate Therapeutics, Aprea Therapeutics. W.G.W. served in a consulting or advisory role for Sanofi; has received research funding from GlaxoSmithKline/Novartis, AbbVie, Genentech, Karyopharm Therapeutics, Pharmacyclics, Acerta Pharma, Gilead Sciences, Janssen, Emergent BioSolutions, Juno Therapeutics, Kite Pharma, Oncternal Therapeutics, Inc, Loxo, Xencor, miRagen, Sunesis Pharmaceuticals, Cyclacel. S.D. declares no competing financial interests.
Correspondence: John C. Byrd, The Ohio State University, 455B OSUCCC, 410 W 12th Ave, Columbus, OH 43210; e-mail: john.byrd@osumc.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal