

In this issue of Blood, 1 report that by simultaneously sampling 2 nodes from distinct anatomical sites in patients with follicular lymphoma (FL), the diverse spectrum of an individual patient’s disease is revealed. Biological studies of FL to date have largely focused on mechanisms and/or prognosticators to account for differences between patients. The current manuscript takes a different approach: examining differences across anatomic sites within an individual patient. The divergent evolution within the same patient helps explain treatment failures and relapse and explains the discordant response to therapy that is often seen in FL.
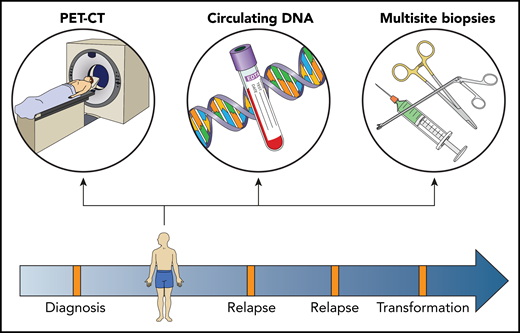
Capturing spatial and dynamic heterogeneity in follicular lymphoma. Sampling of multiple nodal and extrandodal sites, to provide tissue for genomic, transcriptomic, proteomic, and functional assays on the malignant B-cell environment and TME, combined with PET-CT scans and circulating DNA, will provide a comprehensive biological profile. Sequential assays, performed at diagnosis and all subsequent clinical inflection time points, enable the temporal dynamics of site-to-site heterogeneity to be captured. Professional illustration by Patrick Lane, ScEYEnce Studios.
Capturing spatial and dynamic heterogeneity in follicular lymphoma. Sampling of multiple nodal and extrandodal sites, to provide tissue for genomic, transcriptomic, proteomic, and functional assays on the malignant B-cell environment and TME, combined with PET-CT scans and circulating DNA, will provide a comprehensive biological profile. Sequential assays, performed at diagnosis and all subsequent clinical inflection time points, enable the temporal dynamics of site-to-site heterogeneity to be captured. Professional illustration by Patrick Lane, ScEYEnce Studios.
The importance of the findings of Haebe et al is underscored by studies of the spatial heterogeneity in solid organ cancers.2 Biopsies from nonhematologic malignancies taken from distinct body sites demonstrate genetic variability.2 This not only has mechanistic implications in terms of chemoresistance, but also impairs our ability to accurately predict the optimal therapeutic strategy. Likewise, this has major implications for the biomarker driven precision-based oncologic paradigm. In FL, this is perhaps best illustrated by prior observations regarding spatially discordant mutations in EZH2,3 which is a predictive biomarker for the EZH2 inhibitor tazemetostat and an integral component of the clinico-genetic prognosticator M7-FLIPI.4,5 In this paper, Haebe et al find discordance in the expression of other emergent therapeutic targets such as HDAC9 and CD79B.
Most cases of FL show a uniform pattern of closely packed abnormal follicles composed of germinal center B cells. These cells are interspersed within a nonmalignant tumor microenvironment (TME) that consists mostly of T cells. However, in some cases, the architecture of FL within the node is variable, including differences in the extent of immune cell infiltration. Thus, there is intratumoral heterogeneity even within the biopsy site. However, as the mainstay of diagnosis of FL remains histologic assessment of a biopsy from a single site, until now, the understanding of the degree of spatial heterogeneity between noncontiguous diseased areas within an individual patient has been limited.
FL has a diverse genomic landscape, including the presence of different dominant and subdominant malignant B-cell clones.6 A previous study of synchronously acquired paired nodal and non-nodal FL tumors found different patterns of somatic mutations across sites.3 Broadly, their findings showed spatially concordant truncal mutations in early genetic drivers such as CREBBP and KMT2D, whereas spatially discordant variants were also seen involving a diverse range of genes including the cell cycle and B-cell receptor signaling pathways.
Haebe et al expands on previous studies of intratumoral heterogeneity using an elegant multiomic approach to examine the malignant cells and the TME. Immunoglobulin gene rearrangements were used to track individual B-cell clones. In some patients, there was divergent evolution with dominant subclones distinct to anatomic sites. By contrast, other patients demonstrated major subclones shared between the tumor sites, suggesting tumor cell migration. Cases with a high degree of site-to-site phylogenic dissimilarity were associated with higher rates of somatic hypermutation indicating a possible mechanism underpinning these patterns of clonal evolution.
Despite observing instances of site-restricted tumor subclones, the TME was phenotypically relatively uniform across tumor sites. The one notable variation was that the abundance of follicular helper T cells correlated with site-to-site tumor heterogeneity, which reflected tumor CD40 expression.
The apparent homogeneity of the TME across anatomic sites supports the contention that immune-based biomarkers are reliable predictors of clinical behavior independent of biopsy site.7 Because Haebe et al was restricted by the use of cell suspensions, analysis of the TME was limited with respect to intratumoral spatial profiling. This is noteworthy because spatial organization of FL has been shown to result in differential FL biology, immune interactions, and resistance mechanisms to commonly used anticancer agents.8 Furthermore, Haebe et al showed that individual T-cell clones had migrated between anatomical sites, indicating additional complexity of spatial site-to-site heterogeneity within the TME.
The biological basis of heterogeneity in 18F-fluorodeoxyglucose avidity in FL tumors, frequently seen in clinical practice, remains undefined. Here, Haebe et al make some intriguing observations.1 They found a nonsignificant correlative trend between gene expression and the maximum standard uptake value, suggesting that some aspects of site-to-site heterogeneity, perhaps reflecting the malignant B-cell and/or TME compartments, may be reflected by differences in glucose uptake within the diseased nodes. However, because positron emission tomography–computerized tomography (PET-CT) scans were available in only 7 of 10 patients, future investigations incorporating large groups of patients that directly assess glucose uptake within malignant FL B cells and infiltrating T cells must be performed before definitive conclusions can be reached.
Temporal and spatial heterogeneity is an important concept in understanding treatment resistance in FL. Previous studies have concentrated on the notion of evolving tumor cell fitness, with FL transformation typically accompanied by marked expansion of aggressive malignant clones (present at low frequency at diagnosis) because of selection pressures driven by therapeutic interventions.9 In contrast, FL progression generally shows a different pattern of clonal dynamics, with expansion of B-cell clones that are already sizeable at initial presentation. As this study involved biopsies taken at a snapshot in time from previously treated patients, clonal dynamics as a component of site-to-site heterogeneity remains an issue to be addressed in subsequent studies.
Although patient-derived xenografts offer the opportunity to study FL dynamics in a controlled setting, currently such models require engraftment in immunocompromised mice. Crucially, this prevents analysis of the bidirectional relationship that shapes both malignant FL B cells and their TME.10 To address this in humans, temporal studies would do best if they used a holistic approach (see figure), incorporating PET, multiregional tissue sampling, multiomic and functional analysis of FL B cells, and the TME of circulating DNA, all taken sequentially during the course of disease (ie, at diagnosis and all clinical inflection time points). Perhaps the study of Haebe et al not only highlights the limitations of the current single-site biopsy approach but also points the way forward for future studies of heterogeneity in FL.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal