

Key Points
Loncastuximab tesirine demonstrated manageable safety and notable antitumor activity in R/R B-NHL.
A phase 2 study using a dosing regimen based on cumulative safety, pharmacokinetic, and efficacy data from this study has been conducted.

Abstract
The prognosis for patients with relapsed or refractory (R/R) B-cell non-Hodgkin lymphoma (B-NHL) remains poor, with a need for alternatives to current salvage therapies. Loncastuximab tesirine (ADCT-402) is an antibody-drug conjugate comprising a humanized anti-CD19 monoclonal antibody conjugated to a pyrrolobenzodiazepine dimer toxin. Presented here are final results of a phase 1 dose-escalation and dose-expansion study in patients with R/R B-NHL. Objectives were to determine the maximum tolerated dose (MTD) and recommended dose(s) for expansion and evaluate safety, clinical activity, pharmacokinetics, and immunogenicity of loncastuximab tesirine. Overall, 183 patients received loncastuximab tesirine, with 3 + 3 dose escalation at 15 to 200 µg/kg and dose expansion at 120 and 150 µg/kg. Dose-limiting toxicities (all hematologic) were reported in 4 patients. The MTD was not reached, although cumulative toxicity was higher at 200 µg/kg. Hematologic treatment-emergent adverse events were most common, followed by fatigue, nausea, edema, and liver enzyme abnormalities. Overall response rate (ORR) in evaluable patients was 45.6%, including 26.7% complete responses (CRs). ORRs in patients with diffuse large B-cell lymphoma (DLBCL), mantle cell lymphoma, and follicular lymphoma were 42.3%, 46.7%, and 78.6%, respectively. Median duration of response in all patients was 5.4 months and not reached in patients with DLBCL (doses ≥120 µg/kg) who achieved a CR. Loncastuximab tesirine had good stability in serum, notable antitumor activity, and an acceptable safety profile, warranting continued study in B-NHL. The recommended dose for phase 2 was determined as 150 µg/kg every 3 weeks for 2 doses followed by 75 µg/kg every 3 weeks. This trial was registered at www.clinicaltrials.gov as #NCT02669017.
Introduction
B-cell non-Hodgkin lymphoma (B-NHL) includes both aggressive, most commonly diffuse large B-cell lymphoma (DLBCL), and indolent types, most commonly follicular lymphoma (FL).1-4 Approximately 60% of patients with DLBCL can be cured with first-line chemoimmunotherapies.5,6 Options for patients with relapsed or refractory (R/R) DLBCL include salvage chemotherapy with autologous hematopoietic cell transplantation (HCT) or chimeric antigen receptor (CAR) T-cell therapy.7 However, outcomes with salvage therapy for patients who are refractory to treatment or relapse remain poor,8 highlighting the need for new therapeutic options. Indolent forms of B-NHL, such as FL, generally respond to treatment but are infrequently curable, and patients with early relapse have particularly poor outcomes.3 Novel approaches, such as antibody-based treatments, immune checkpoint inhibitors, and small-molecule inhibitors, could improve outcomes for those with R/R B-NHL and/or reduce toxicities seen with standard treatments.2
Loncastuximab tesirine (ADCT-402) is an antibody-drug conjugate (ADC) comprising a humanized anti-CD19 monoclonal antibody stochastically conjugated through a cathepsin-cleavable valine-alanine linker to a pyrrolobenzodiazepine (PBD) dimer toxin, SG3199.9 CD19 is a suitable target for immunotherapy for B-NHL because it is normally expressed during B-cell development, but only after B-lineage commitment and thus not on hematopoietic stem cells,10,11 and CD19 expression is lost during terminal plasma cell differentiation but maintained in hematologic B-cell malignancies.10-12
PBD dimers are sequence-selective, nondistorting, and potent cytotoxic DNA crosslinking agents.13-15 The interstrand crosslinks formed between DNA in the minor groove and PBD are relatively nondistorting of the DNA structure, preventing detection by repair mechanisms and seeming to contribute to the persistence and potent biologic activity of PBD in cells.15,16 Preclinically, loncastuximab tesirine showed highly targeted antitumor effects in vitro and in vivo with DNA-PBD crosslinks persisting for up to 36 hours.9
Data from the dose-escalation part (part 1) of the first-in-human study of loncastuximab tesirine in adults with R/R B-NHL demonstrated promising single-agent activity and acceptable safety in patients with R/R B-NHL.17 Here, we report results for the full study population in part 1 and part 2 (dose expansion).
Methods
Patients
Adults (age ≥18 years) with histologically confirmed R/R B-NHL (World Health Organization 2008 classification18 ) who were intolerant to established therapy, for whom established therapy had failed, or for whom no other treatment options were available in the opinion of the investigator were eligible to participate. Inclusion and exclusion criteria are presented in the supplemental Data (available on the Blood Web site).
The clinical study was performed per the International Council for Harmonisation good clinical practice guidelines and the ethical principles of the Declaration of Helsinki and was approved by each institutional review board. All patients provided written informed consent.
Study design and treatment
This phase 1, open-label, 2-part (dose escalation [part 1] and dose expansion [part 2]) study of loncastuximab tesirine monotherapy was conducted in patients with R/R B-NHL at 11 centers in 3 countries (United States, United Kingdom, and Italy; enrollment from 9 March 2016 to 8 May 2018). Primary objectives of part 1 were to evaluate safety and tolerability of loncastuximab tesirine in R/R B-NHL and determine the maximum tolerated dose (MTD) and recommended dose(s) for expansion (part 2). Primary objectives for part 2 were to evaluate safety and tolerability at the recommended dose(s). Secondary objectives included evaluation of antitumor activity; characterization of exposure to total antibody, PBD-conjugated antibody, and free warhead at different doses and cycles using standard pharmacokinetic (PK) parameters; and evaluation of induction of antidrug antibodies (ADAs) to loncastuximab tesirine. Exploratory assessments included evaluation of changes in peripheral white blood cell (WBC) counts and CD markers and correlations between baseline CD19 levels in archival tumor tissue and PK and clinical activity of loncastuximab tesirine.
Loncastuximab tesirine was administered by IV infusion over 60 minutes once every 3 weeks (day 1 of each 21-day cycle). In part 1, patients were assigned to doses using a 3 + 3 dose-escalation design (starting dose, 15 μg/kg every 3 weeks), overseen by a dose-escalation steering committee. No intrapatient dose escalation was permitted. In part 2, patients were assigned to recommended dose level(s) and regimen(s) of loncastuximab tesirine identified in part 1, based on safety, efficacy, and PK data, with ongoing dose-escalation steering committee–directed enrollment to enable evaluation of different dosing regimens for doses identified for further evaluation in part 1. No formal sample size justification was performed, because the primary objective was to evaluate safety. Treatment administration is described in the supplemental Data.
Dose-limiting toxicities (DLTs) were defined as described in the supplemental Data during cycle 1 in part 1 (DLT observation period), except when events were clearly due to underlying disease or extraneous causes. Based on the 3 + 3 design, the MTD was the highest dose level at which 0 of the first 3 patients treated or ≤1 of the first 6 patients treated had a DLT during cycle 1 of part 1.
Patients received loncastuximab tesirine until disease progression, unacceptable toxicity, initiation of new anticancer treatment, or withdrawal from the study. Patients who discontinued treatment for a reason other than progressive disease (PD) were followed every 12 weeks until PD or initiation of new anticancer treatment, and patients were followed for survival for ≤12 months after last dose of study drug.
Assessments
Safety assessments included adverse events (AEs), serious AEs, DLTs, periodic 12-lead electrocardiograms, physical examinations, vital signs, Eastern Cooperative Oncology Group performance status, and laboratory tests (hematology, coagulation panel, biochemistry, pregnancy testing [in women of childbearing potential], and urinalysis). AEs were classified using the Medical Dictionary for Regulatory Activities (version 22.0). Treatment-emergent AEs (TEAEs) were defined as AEs that began or worsened during or after the first dose of study drug until 12 weeks after the last dose or until the initiation of new anticancer treatment. TEAEs were graded per the National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.0).
Antitumor activity measures were overall response rate (ORR), duration of response (DOR), overall survival (OS), and progression-free survival (PFS). Disease assessments occurred every other cycle for the first 2 evaluations (at 6 weeks [end of cycle 2 ± 1 week], at 12 weeks [end of cycle 4 ± 1 week]), and then every third cycle (end of cycles 7, 10, and so on) until PD, or more frequently if indicated clinically, according to local site imaging requirements (positron emission tomography-computed tomography or computed tomography), with the same method used for all assessments in each patient. Investigators classified patients’ responses to treatment as complete response (CR), partial response (PR), stable disease, or PD (2014 Lugano classification).19
Blood samples for PK analysis were collected as described in supplemental Table 1. Blood samples for ADA analysis were collected on days 1 (preinfusion) and 21 of each cycle, at end of treatment, and during follow-up. Validated bioanalytic methods were used to determine standard PK parameters for loncastuximab tesirine total antibody, PBD-conjugated antibody, and free warhead SG3199 and determine ADAs to loncastuximab tesirine. Exploratory assessments included immunohistochemistry of archival/pretreatment tumor samples for CD19 protein expression and flow cytometry for peripheral WBC changes.
Statistical analysis
Safety was analyzed in patients who received study drug, as were DLTs in all patients who completed at least 1 cycle in part 1 or who discontinued before cycle end but had complete DLT information. Evaluation set descriptions are provided in the supplemental Data.
Descriptive statistics and data were used to report end points. Data are reported for each patient’s starting dose of loncastuximab tesirine. DOR, PFS, and OS were estimated using the Kaplan-Meier method with censoring. Efficacy end points were analyzed in all patients with B-NHL and by histology. ORR was also analyzed by predefined subgroups. Noncompartmental analysis was used to determine PK parameters. Serum concentrations of PBD-conjugated antibody were used in population PK modeling to obtain individual patient metrics of drug exposure. Relationships between exposure and TEAEs were analyzed and binomial logistic regression performed to predict probability of events for a given degree of exposure where event severity had an apparent relationship with drug exposure (supplemental Data). Relationships between CD19 expression in pretreatment/archival tumor tissue samples and exposure to PBD-conjugated antibody in cycle 1 and correlations between PK exposure and peripheral CD19+ B cells were evaluated using linear regression analysis.
Results
Patient disposition and characteristics
In total, 183 patients received loncastuximab tesirine. In part 1, 88 patients were treated with doses of 15 to 200 μg/kg every 3 weeks (15, 30, or 60 μg/kg, n = 4 each; 90 μg/kg, n = 5; 120 μg/kg, n = 16; 150 μg/kg, n = 19; 200 μg/kg, n = 36). Cumulative toxicity observed at 200 μg/kg every 3 weeks led to a protocol amendment, with 22 patients assigned to 200 μg/kg receiving loncastuximab tesirine every 6 weeks during part 1.
Based on an increase in cumulative toxicities at 200 μg/kg and evidence of activity at 120 and 150 μg/kg during part 1, doses of 120 μg/kg every 3 weeks and 150 µg/kg every 3 weeks were selected for part 2, with some patients in the 150 µg/kg group reducing their dose to 75 μg/kg every 3 weeks after 3 cycles. In part 2, 26 patients received loncastuximab tesirine at 120 μg/kg (parts 1 + 2, n = 42), and 69 patients received 150 μg/kg (parts 1 + 2, n = 88). Patient disposition is shown in Figure 1. The most common reason for treatment discontinuation was PD (83 [45.4%] of 183); the most common reason for study discontinuation was death (111 [60.7%] of 183).

Patient disposition.aA patient was considered to have completed the study after 12 months posttreatment follow-up data were obtained.
Patient disposition.aA patient was considered to have completed the study after 12 months posttreatment follow-up data were obtained.
Baseline characteristics for all patients and those with DLBCL are presented in Table 1; most patients had DLBCL (139 [76.0%] of 183), 15 (8.2%) had mantle cell lymphoma (MCL), 14 (7.7%) had FL, and 15 (8.2%) had B-NHL of other histologies. Patients had received a median of 3 prior lines of systemic therapy (range, 1-13 lines); 42 (23%) had received prior HCT, and 3 (1.6%) had received prior CAR T-cell therapy. Forty-three patients (23.5%) were primary refractory and 109 (59.6%) were refractory to their most recent systemic therapy.
Baseline demographic and clinical characteristics of patients with B-NHL who received loncastuximab tesirine (safety analysis set)
| Characteristic | n (%) | |
|---|---|---|
| All patients with B-NHL (N = 183) | Patients with DLBCL (n = 139) | |
| Sex | ||
| Female | 69 (37.7) | 59 (42.2) |
| Male | 114 (62.3) | 80 (57.6) |
| Age, y | ||
| Median | 63.0 | 63.0 |
| Range | 20-87 | 20-86 |
| ECOG score | ||
| 0-1 | 160 (87.4) | 119 (85.6) |
| 2 | 21 (11.5) | 18 (12.9) |
| 3 | 2 (1.1) | 2 (1.4) |
| B-NHL subtype | ||
| DLBCL group* | ||
| Double hit (MYC plus BCL-2 and/or BCL-6 rearrangement) | 20 (14.4) | |
| Triple hit (MYC plus BCL-2 and BCL-6 rearrangement) | 3 (2.2) | |
| Transformed | 37 (26.6) | |
| MCL | 15 (8.2) | — |
| FL | 14 (7.7)† | — |
| CLL | 6 (3.3) | — |
| Marginal zone B-cell lymphoma | 6 (3.3) | — |
| Burkitt lymphoma | 1 (0.5) | — |
| Waldenström macroglobulinemia | 1 (0.5) | — |
| Other | 1 (0.5)‡ | — |
| No. of lines of prior systemic therapy | ||
| Median | 3 | 3 |
| Range | 1-13 | 1-10 |
| First-line prior systemic therapy response | ||
| Relapsed after initial response | 115 (62.8) | 90 (64.7) |
| Refractory to first-line therapy | 43 (23.5) | 30 (21.6) |
| Last-line prior systemic therapy response | ||
| Relapsed after initial response | 66 (36.1) | 49 (35.3) |
| Refractory to last therapy line | 109 (59.6) | 83 (59.7) |
| Prior HCT | ||
| Autologous | 31 (16.9) | 22 (15.8) |
| Allogeneic | 5 (2.7) | 2 (1.4) |
| Both | 4 (2.2) | 2 (1.4) |
| Other§ | 2 (1.1) | 1 (0.7) |
| Prior CAR T-cell therapy | ||
| Yes | 3 (1.6) | 2 (1.4) |
| No | 180 (98.4) | 137 (98.6) |
| Serum LDH, U/L | ||
| Median | 323.0 | |
| Range | 109-9348 | |
| Characteristic | n (%) | |
|---|---|---|
| All patients with B-NHL (N = 183) | Patients with DLBCL (n = 139) | |
| Sex | ||
| Female | 69 (37.7) | 59 (42.2) |
| Male | 114 (62.3) | 80 (57.6) |
| Age, y | ||
| Median | 63.0 | 63.0 |
| Range | 20-87 | 20-86 |
| ECOG score | ||
| 0-1 | 160 (87.4) | 119 (85.6) |
| 2 | 21 (11.5) | 18 (12.9) |
| 3 | 2 (1.1) | 2 (1.4) |
| B-NHL subtype | ||
| DLBCL group* | ||
| Double hit (MYC plus BCL-2 and/or BCL-6 rearrangement) | 20 (14.4) | |
| Triple hit (MYC plus BCL-2 and BCL-6 rearrangement) | 3 (2.2) | |
| Transformed | 37 (26.6) | |
| MCL | 15 (8.2) | — |
| FL | 14 (7.7)† | — |
| CLL | 6 (3.3) | — |
| Marginal zone B-cell lymphoma | 6 (3.3) | — |
| Burkitt lymphoma | 1 (0.5) | — |
| Waldenström macroglobulinemia | 1 (0.5) | — |
| Other | 1 (0.5)‡ | — |
| No. of lines of prior systemic therapy | ||
| Median | 3 | 3 |
| Range | 1-13 | 1-10 |
| First-line prior systemic therapy response | ||
| Relapsed after initial response | 115 (62.8) | 90 (64.7) |
| Refractory to first-line therapy | 43 (23.5) | 30 (21.6) |
| Last-line prior systemic therapy response | ||
| Relapsed after initial response | 66 (36.1) | 49 (35.3) |
| Refractory to last therapy line | 109 (59.6) | 83 (59.7) |
| Prior HCT | ||
| Autologous | 31 (16.9) | 22 (15.8) |
| Allogeneic | 5 (2.7) | 2 (1.4) |
| Both | 4 (2.2) | 2 (1.4) |
| Other§ | 2 (1.1) | 1 (0.7) |
| Prior CAR T-cell therapy | ||
| Yes | 3 (1.6) | 2 (1.4) |
| No | 180 (98.4) | 137 (98.6) |
| Serum LDH, U/L | ||
| Median | 323.0 | |
| Range | 109-9348 | |
CLL, chronic lymphocytic leukemia; ECOG, Eastern Oncology Cooperative Group; LDH, lactate dehydrogenase; U/L, upper/lower.
DLBCL subtypes comprised DLBCL (n = 134), high-grade B-cell lymphoma (BCL; n = 2), aggressive BCL with features intermediate between DLBCL and Burkitt lymphoma (n = 1), mediastinal BCL (thymic large BCL; n = 1), and primary mediastinal BCL (n = 1). In the DLBCL category, transformed disease comprised FL (n = 26), marginal zone B-cell lymphoma (n = 2), lymphoplasmacytic lymphoma (n = 1), nodular lymphocyte-predominant Hodgkin lymphoma (n = 2), and Richter’s transformation (n = 6).
One patient with FL also had CLL/small lymphocytic lymphoma recurrence.
This patient had a history of DLBCL and was enrolled based on imaging consistent with recurrence. The patient was subsequently biopsied after enrollment, and the lesion determined to be sarcoid.
One patient with DLBCL underwent peripheral stem cell harvest transplantation, and 1 patient with FL underwent double cord transplantation.
Safety
Safety and DLT analysis sets comprised 183 and 73 patients, respectively. Patients received a median of 2 doses (range, 1-24 doses) of loncastuximab tesirine, with a median weight-adjusted dose per cycle of 129.9 μg/kg (range, 14.6-204.4 μg/kg) for a median duration of 64 days (range, 22-532 days).
Four patients experienced DLTs during part 1: grade 4 thrombocytopenia in 1 patient receiving 120 μg/kg (1 of 16), grade 3 febrile neutropenia in 1 patient receiving 150 μg/kg (1 of 16), and grade 4 thrombocytopenia in 2 patients receiving 200 μg/kg (2 of 25). The MTD was not reached.
In the safety analysis set, 181 patients (98.9%) had at least 1 TEAE. TEAEs (≥10% of patients; Table 2) were consistent with those reported previously for loncastuximab tesirine.17 Hematologic TEAEs were common, including platelet count decreased, neutrophil count decreased (both based on laboratory abnormality reporting), and anemia. Fatigue was the most common nonhematologic TEAE (78 [42.6%] of 183), followed by nausea (59 [32.2%] of 183), peripheral edema (58 [31.7%] of 183), and GGT increased (57 [31.1%] of 183). Accumulating toxicity was apparent with loncastuximab tesirine at 200 μg/kg, with many TEAEs more common in the 200 μg/kg group than in lower-dose groups, including hematologic abnormalities, peripheral edema, and liver test abnormalities (Table 2).
All-grade TEAEs reported in ≥10% of patients with B-NHL who received loncastuximab tesirine in order of incidence by system order class (safety analysis set)
| TEAE | n (%) | ||||
|---|---|---|---|---|---|
| ≤90 µg/kg (n = 17) | 120 µg/kg (n = 42) | 150 µg/kg (n = 88) | 200 µg/kg (n = 36) | Total (N = 183) | |
| Any | 16 (94.1) | 42 (100) | 87 (98.9) | 36 (100) | 181 (98.9) |
| Hematologic | |||||
| Platelet count decreased* | 11 (64.7) | 28 (68.3) | 62 (71.3) | 27 (77.1) | 128 (71.1) |
| Neutrophil count decreased* | 10 (58.8) | 21 (51.2) | 50 (58.1) | 25 (71.4) | 106 (59.2) |
| Anemia | 4 (23.5) | 10 (23.8) | 32 (36.4) | 14 (38.9) | 60 (32.8) |
| WBC count decreased | 0 | 7 (16.7) | 6 (6.8) | 9 (25.0) | 22 (12.0) |
| Nonhematologic | |||||
| General disorders and administration site conditions | |||||
| Fatigue | 7 (41.2) | 22 (52.4) | 33 (37.5) | 16 (44.4) | 78 (42.6) |
| Edema peripheral | 1 (5.9) | 12 (28.6) | 31 (35.2) | 14 (38.9) | 58 (31.7) |
| Pyrexia | 2 (11.8) | 7 (16.7) | 13 (14.8) | 11 (30.6) | 33 (18.0) |
| Gastrointestinal disorders | |||||
| Nausea | 3 (17.6) | 12 (28.6) | 28 (31.8) | 16 (44.4) | 59 (32.2) |
| Constipation | 2 (11.8) | 12 (28.6) | 20 (22.7) | 6 (16.7) | 40 (21.9) |
| Vomiting | 1 (5.9) | 7 (16.7) | 17 (19.3) | 7 (19.4) | 32 (17.5) |
| Abdominal pain | 1 (5.9) | 9 (21.4) | 12 (13.6) | 7 (19.4) | 29 (15.8) |
| Diarrhea | 2 (11.8) | 5 (11.9) | 16 (18.2) | 5 (13.9) | 28 (15.3) |
| Investigations | |||||
| GGT increased | 5 (29.4) | 13 (31.0) | 22 (25.0) | 17 (47.2) | 57 (31.1) |
| Blood ALP increased | 4 (23.5) | 6 (14.3) | 18 (20.5) | 9 (25.0) | 37 (20.2) |
| AST increased | 3 (17.6) | 5 (11.9) | 15 (17.0) | 11 (30.6) | 34 (18.6) |
| ALT increased | 3 (17.6) | 6 (14.3) | 14 (15.9) | 9 (25.0) | 32 (17.5) |
| Skin and subcutaneous tissue disorders | |||||
| Rash | 2 (11.8) | 7 (16.7) | 27 (30.7) | 9 (25.0) | 45 (24.6) |
| Erythema | 1 (5.9) | 5 (11.9) | 11 (12.5) | 4 (11.1) | 21 (11.5) |
| Pruritus | 2 (11.8) | 4 (9.5) | 7 (8.0) | 7 (19.4) | 20 (10.9) |
| Rash maculopapular | 3 (17.6) | 4 (9.5) | 7 (8.0) | 5 (13.9) | 19 (10.4) |
| Metabolism and nutrition disorders | |||||
| Decreased appetite | 2 (11.8) | 7 (16.7) | 13 (14.8) | 12 (33.3) | 34 (18.6) |
| Hypokalemia | 1 (5.9) | 3 (7.1) | 15 (17.0) | 4 (11.1) | 23 (12.6) |
| Hyperglycemia | 1 (5.9) | 3 (7.1) | 10 (11.4) | 5 (13.9) | 19 (10.4) |
| Respiratory, thoracic, and mediastinal disorders | |||||
| Dyspnea | 1 (5.9) | 11 (26.2) | 21 (23.9) | 8 (22.2) | 41 (22.4) |
| Pleural effusion | 2 (11.8) | 10 (23.8) | 19 (21.6) | 8 (22.2) | 39 (21.3) |
| Cough | 0 | 10 (23.8) | 16 (18.2) | 8 (22.2) | 34 (18.6) |
| Nervous system disorders | |||||
| Dizziness | 1 (5.9) | 6 (14.3) | 9 (10.2) | 4 (11.1) | 20 (10.9) |
| TEAE | n (%) | ||||
|---|---|---|---|---|---|
| ≤90 µg/kg (n = 17) | 120 µg/kg (n = 42) | 150 µg/kg (n = 88) | 200 µg/kg (n = 36) | Total (N = 183) | |
| Any | 16 (94.1) | 42 (100) | 87 (98.9) | 36 (100) | 181 (98.9) |
| Hematologic | |||||
| Platelet count decreased* | 11 (64.7) | 28 (68.3) | 62 (71.3) | 27 (77.1) | 128 (71.1) |
| Neutrophil count decreased* | 10 (58.8) | 21 (51.2) | 50 (58.1) | 25 (71.4) | 106 (59.2) |
| Anemia | 4 (23.5) | 10 (23.8) | 32 (36.4) | 14 (38.9) | 60 (32.8) |
| WBC count decreased | 0 | 7 (16.7) | 6 (6.8) | 9 (25.0) | 22 (12.0) |
| Nonhematologic | |||||
| General disorders and administration site conditions | |||||
| Fatigue | 7 (41.2) | 22 (52.4) | 33 (37.5) | 16 (44.4) | 78 (42.6) |
| Edema peripheral | 1 (5.9) | 12 (28.6) | 31 (35.2) | 14 (38.9) | 58 (31.7) |
| Pyrexia | 2 (11.8) | 7 (16.7) | 13 (14.8) | 11 (30.6) | 33 (18.0) |
| Gastrointestinal disorders | |||||
| Nausea | 3 (17.6) | 12 (28.6) | 28 (31.8) | 16 (44.4) | 59 (32.2) |
| Constipation | 2 (11.8) | 12 (28.6) | 20 (22.7) | 6 (16.7) | 40 (21.9) |
| Vomiting | 1 (5.9) | 7 (16.7) | 17 (19.3) | 7 (19.4) | 32 (17.5) |
| Abdominal pain | 1 (5.9) | 9 (21.4) | 12 (13.6) | 7 (19.4) | 29 (15.8) |
| Diarrhea | 2 (11.8) | 5 (11.9) | 16 (18.2) | 5 (13.9) | 28 (15.3) |
| Investigations | |||||
| GGT increased | 5 (29.4) | 13 (31.0) | 22 (25.0) | 17 (47.2) | 57 (31.1) |
| Blood ALP increased | 4 (23.5) | 6 (14.3) | 18 (20.5) | 9 (25.0) | 37 (20.2) |
| AST increased | 3 (17.6) | 5 (11.9) | 15 (17.0) | 11 (30.6) | 34 (18.6) |
| ALT increased | 3 (17.6) | 6 (14.3) | 14 (15.9) | 9 (25.0) | 32 (17.5) |
| Skin and subcutaneous tissue disorders | |||||
| Rash | 2 (11.8) | 7 (16.7) | 27 (30.7) | 9 (25.0) | 45 (24.6) |
| Erythema | 1 (5.9) | 5 (11.9) | 11 (12.5) | 4 (11.1) | 21 (11.5) |
| Pruritus | 2 (11.8) | 4 (9.5) | 7 (8.0) | 7 (19.4) | 20 (10.9) |
| Rash maculopapular | 3 (17.6) | 4 (9.5) | 7 (8.0) | 5 (13.9) | 19 (10.4) |
| Metabolism and nutrition disorders | |||||
| Decreased appetite | 2 (11.8) | 7 (16.7) | 13 (14.8) | 12 (33.3) | 34 (18.6) |
| Hypokalemia | 1 (5.9) | 3 (7.1) | 15 (17.0) | 4 (11.1) | 23 (12.6) |
| Hyperglycemia | 1 (5.9) | 3 (7.1) | 10 (11.4) | 5 (13.9) | 19 (10.4) |
| Respiratory, thoracic, and mediastinal disorders | |||||
| Dyspnea | 1 (5.9) | 11 (26.2) | 21 (23.9) | 8 (22.2) | 41 (22.4) |
| Pleural effusion | 2 (11.8) | 10 (23.8) | 19 (21.6) | 8 (22.2) | 39 (21.3) |
| Cough | 0 | 10 (23.8) | 16 (18.2) | 8 (22.2) | 34 (18.6) |
| Nervous system disorders | |||||
| Dizziness | 1 (5.9) | 6 (14.3) | 9 (10.2) | 4 (11.1) | 20 (10.9) |
ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, γ-glutamyltransferase.
Platelet count decreased and neutrophil count decreased are based on laboratory abnormality reporting and are reported out of number of patients with postbaseline test values; data for 4 patients (1 at 120 µg/kg, 2 at 150 µg/kg, and 1 at 200 µg/kg) were missing for neutrophil count decreased, and data for 3 patients (1 each at 120, 150, and 200 µg/kg) were missing for platelet count decreased.
Laboratory values for platelet, neutrophil, and GGT levels are presented in supplemental Figure 2. Generally, platelet counts followed a pattern of decrease and recovery, which was most pronounced at 200 µg/kg, with limited partial platelet recovery reflective of accumulating toxicity at this dose. Grade 3/4 platelet count decreases were most common during the first 2 cycles; patients with prolonged events had treatment withdrawn. Neutrophil counts decreased from baseline to cycle 1, day 15; additional decreases were not apparent, except for slight decreases at 90 μg/kg up to cycle 3, day 1. GGT levels seemed to increase over time, particularly at higher doses.
Skin- or nail-related toxicities were reported in 98 patients (53.6%; most commonly rash [45 (24.6%) of 183], erythema [21 (11.5%) of 183], pruritus [20 (10.9%) of 183], and maculopapular rash [19 (10.4%) of 183]) and were generally mild to moderate and reversible but were sometimes prolonged. Rash was most common in sun exposed areas, and most affected patients continued treatment as planned; a minority were managed with dose delays (1.6% each of patients with rash and maculopapular rash), and 2 patients (1.1%) discontinued treatment. Edema or effusion was reported in 86 patients (47.0%), including peripheral edema in 58 (31.7%) and pleural effusion in 39 (21.3%). These events generally occurred after at least 2 cycles. Most patients continued treatment, although some required dose delays. Introducing dexamethasone premedication reduced incidence of edema or effusion in part 2 (120 µg/kg, 34.6%; 150 µg/kg, 47.8%) vs part 1 (120 µg/kg, 68.8%; 150 µg/kg, 63.2%). One patient who received loncastuximab tesirine at 120 μg/kg had a grade 2 infusion-related reaction during cycle 1 on day 1 in part 2 that resolved on the same day; dosing was not modified, and the patient received 8 additional cycles of loncastuximab tesirine.
Grade ≥3 TEAEs (≥5% of all patients) are listed in Table 3. Grade ≥3 TEAEs were reported in 141 patients (77%), most commonly hematologic or liver test abnormalities and hypokalemia. Consistent with the overall pattern of TEAEs, several grade ≥3 TEAEs were more common with loncastuximab tesirine at 200 μg/kg than at lower doses, including GGT increased, neutrophil count decreased, and platelet count decreased.
Grade ≥3 TEAEs reported in ≥5% of patients with B-NHL who received loncastuximab tesirine (safety analysis set)
| TEAE | n (%) | ||||
|---|---|---|---|---|---|
| ≤90 µg/kg (n = 17) | 120 µg/kg (n = 42) | 150 µg/kg (n = 88) | 200 µg/kg (n = 36) | Total (N = 183) | |
| Any | 9 (52.9) | 32 (76.2) | 69 (78.4) | 31 (86.1) | 141 (77.0) |
| Neutrophil count decreased* | 6 (35.3%) | 12 (29.3) | 35 (40.7) | 18 (51.4) | 71 (39.7) |
| Platelet count decreased* | 1 (5.9%) | 7 (17.1) | 25 (28.7) | 15 (42.9) | 48 (26.7) |
| GGT increased | 4 (23.5) | 9 (21.4) | 15 (17.0) | 11 (30.6) | 39 (21.3) |
| Anemia | 3 (17.6) | 4 (9.5) | 16 (18.2) | 5 (13.9) | 28 (15.3) |
| Blood ALP increased | 4 (23.5) | 3 (7.1) | 3 (3.4) | 2 (5.6) | 12 (6.6) |
| Lymphocyte count decreased | 0 | 4 (9.5) | 6 (6.8) | 2 (5.6) | 12 (6.6) |
| PD | 0 | 2 (4.8) | 9 (10.2) | 0 | 11 (6.0) |
| Febrile neutropenia | 1 (5.9) | 2 (4.8) | 6 (6.8) | 1 (2.8) | 10 (5.5) |
| Hypokalemia | 0 | 0 | 8 (9.1) | 2 (5.6) | 10 (5.5) |
| TEAE | n (%) | ||||
|---|---|---|---|---|---|
| ≤90 µg/kg (n = 17) | 120 µg/kg (n = 42) | 150 µg/kg (n = 88) | 200 µg/kg (n = 36) | Total (N = 183) | |
| Any | 9 (52.9) | 32 (76.2) | 69 (78.4) | 31 (86.1) | 141 (77.0) |
| Neutrophil count decreased* | 6 (35.3%) | 12 (29.3) | 35 (40.7) | 18 (51.4) | 71 (39.7) |
| Platelet count decreased* | 1 (5.9%) | 7 (17.1) | 25 (28.7) | 15 (42.9) | 48 (26.7) |
| GGT increased | 4 (23.5) | 9 (21.4) | 15 (17.0) | 11 (30.6) | 39 (21.3) |
| Anemia | 3 (17.6) | 4 (9.5) | 16 (18.2) | 5 (13.9) | 28 (15.3) |
| Blood ALP increased | 4 (23.5) | 3 (7.1) | 3 (3.4) | 2 (5.6) | 12 (6.6) |
| Lymphocyte count decreased | 0 | 4 (9.5) | 6 (6.8) | 2 (5.6) | 12 (6.6) |
| PD | 0 | 2 (4.8) | 9 (10.2) | 0 | 11 (6.0) |
| Febrile neutropenia | 1 (5.9) | 2 (4.8) | 6 (6.8) | 1 (2.8) | 10 (5.5) |
| Hypokalemia | 0 | 0 | 8 (9.1) | 2 (5.6) | 10 (5.5) |
ALP, alkaline phosphatase.
Platelet count decreased and neutrophil count decreased are based on laboratory abnormality reporting; data for 4 patients (1 at 120 µg/kg, 2 at 150 µg/kg, and 1 at 200 µg/kg) were missing for neutrophil count decreased, and data for 3 patients (1 each at 120, 150, and 200 µg/kg) were missing for platelet count decreased.
At least 1 serious TEAE was reported in 85 patients (46.4%). Excluding PD, the most common serious TEAEs were febrile neutropenia (10 [5.5%] of 183), pyrexia and pleural effusion (7 [3.8% each] of 183), dyspnea (6 [3.3%] of 183), sepsis (5 [2.7%] of 183), and abdominal pain (4 [2.2%] of 183). Thirty-five patients (19.1%) had TEAEs with a fatal outcome during the study, most commonly (20 of 35) as a result of progression of underlying B-NHL; 6 were considered treatment related, all of which were infections. The pattern of TEAEs among patients with DLBCL was generally similar to that in all patients.
Dose delays of ≤21 days could be used to manage toxicities per protocol, and 68 patients (37.2%) had dose delays because of TEAEs, most commonly (≥5% of patients) GGT increased (19 [10.4%] of 183) and neutropenia (10 [5.5%] of 183). Eleven patients (6.0%) had dose reductions because of TEAEs, and 35 (19.1%) had TEAEs leading to treatment discontinuation, most commonly because of GGT increased (7 patients [3.8%]), followed by thrombocytopenia (5 patients [2.7%]). Few patients with DLBCL (<15%, all doses) had dose modifications (treatment discontinuation, delay, or dose reduction) during the first 2 cycles. The probability of a dose modification increased to ∼30% and ∼50% at the third and fourth doses, respectively.
Antitumor activity
ORR in all patients with B-NHL (180 evaluable) was 45.6% (95% confidence interval [CI], 38.1% to 53.1%), including 48 CRs (26.7%) and 34 PRs (18.9%).
ORR by histology (Table 4) was 42.3% (95% CI, 33.9% to 51.1%) in patients with DLBCL (137 evaluable), 46.7% (95% CI, 21.3% to 73.4%) in patients with MCL (15 evaluable), and 78.6% (95% CI, 49.2% to 95.3%) in patients with FL (14 evaluable). ORR by loncastuximab tesirine dose is shown in supplemental Table 2; ORR for 15 to 90 μg/kg doses was 29.4% compared with 47.2% for 120 to 200 μg/kg doses.
ORRs in B-NHL subgroups treated with loncastuximab tesirine doses 15 to 200 µg (efficacy analysis set)
| n (%) | |||
|---|---|---|---|
| DLBCL (n = 137) | MCL (n = 15) | FL (n = 14) | |
| ORR | 58 (42.3) | 7 (46.7) | 11 (78.6) |
| 95% CI | 33.9-51.1 | 21.3-73.4 | 49.2-95.3 |
| CR | 32 (23.4) | 5 (33.3) | 9 (64.3) |
| PR | 26 (19.0) | 2 (13.3) | 2 (14.3) |
| n (%) | |||
|---|---|---|---|
| DLBCL (n = 137) | MCL (n = 15) | FL (n = 14) | |
| ORR | 58 (42.3) | 7 (46.7) | 11 (78.6) |
| 95% CI | 33.9-51.1 | 21.3-73.4 | 49.2-95.3 |
| CR | 32 (23.4) | 5 (33.3) | 9 (64.3) |
| PR | 26 (19.0) | 2 (13.3) | 2 (14.3) |
Median time to tumor response for all patients with B-NHL who achieved CR or PR was 43.0 days (range, 31-323 days); best percent change from baseline in tumor size is shown by dose and histology in Figure 2.
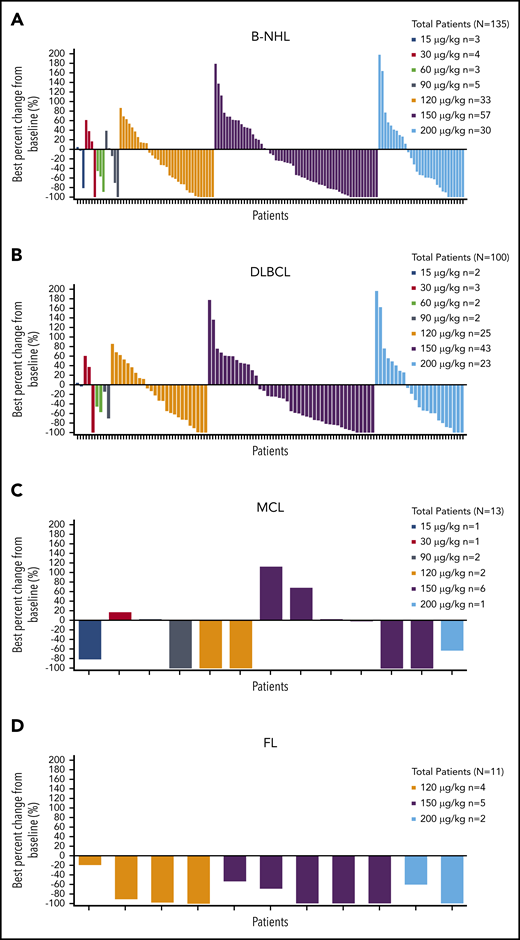
Best percent change from baseline in tumor size by dose. Patients with B-NHL (A), DLBCL (B), MCL (C), and FL (D).
Best percent change from baseline in tumor size by dose. Patients with B-NHL (A), DLBCL (B), MCL (C), and FL (D).
Median DOR with loncastuximab tesirine in all patients with B-NHL was 5.4 months (95% CI, 4.0 months to not reached); it was 4.5 months (95% CI, 3.9-9.5 months) in patients with DLBCL and not reached in patients with MCL or FL (Figure 3A). Similar DORs were achieved in patients with DLBCL receiving 120, 150, and 200 μg/kg doses (Figure 3B). Median DOR was not reached in patients with DLBCL (doses ≥120 µg/kg) who achieved CR (Figure 3C).
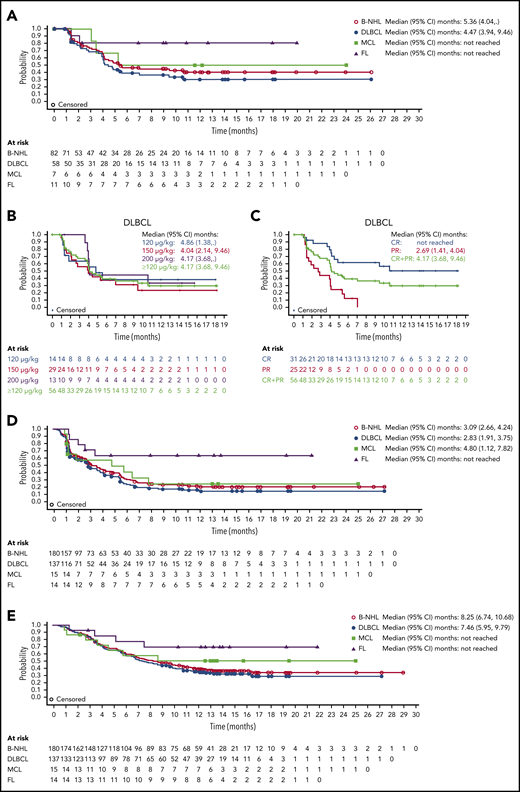
DOR to loncastuximab tesirine, PFS, and OS. DOR by B-NHL subtype (A), for patients with DLBCL by dose (B), and for patients with DLBCL by response (C). (D) PFS for all patients with B-NHL and those with DLBCL, MCL, and FL. (E) OS for all patients with B-NHL and those with DLBCL, MCL, and FL.
DOR to loncastuximab tesirine, PFS, and OS. DOR by B-NHL subtype (A), for patients with DLBCL by dose (B), and for patients with DLBCL by response (C). (D) PFS for all patients with B-NHL and those with DLBCL, MCL, and FL. (E) OS for all patients with B-NHL and those with DLBCL, MCL, and FL.
Median PFS was 3.1 months (95% CI, 2.7-4.2 months) in all patients with B-NHL, 2.8 months (95% CI, 1.9-3.8 months) in patients with DLBCL, 4.8 months (95% CI, 1.1-7.8 months) in patients with MCL, and could not be determined in those with FL because of the low number of events (Figure 3D). Median OS was 8.3 months (95% CI, 6.7-10.7 months) in all patients with B-NHL, 7.5 months (95% CI, 6.0-9.8 months) in patients with DLBCL, and not reached in patients with MCL or FL because of the low number of events (Figure 3E).
A total of 96 patients (52.5%) received subsequent anticancer treatment after receiving loncastuximab tesirine, most commonly systemic therapy (n = 66), radiotherapy (n = 13), and HCT (n = 12). One patient had subsequent CAR T-cell therapy (n = 1).
ORRs for subgroups of patients with DLBCL with high-risk characteristics (supplemental Table 3) were noteworthy in patients age ≥75 years (55.6%), and responses were observed in other difficult-to-treat populations, including patients refractory to first- or last-line therapy (23.3% and 35.8%, respectively) and those with double- or triple-hit lymphoma (21.7%).
PKs
The numbers of patients with sufficient data for PK analysis of PBD-conjugated antibody, total antibody, and free warhead SG3199 were 161, 160, and 37, respectively, across all every-3-week dosing regimens. PKs of loncastuximab tesirine administered every 3 weeks during cycles 1 and 2 are shown in supplemental Tables 4 and 5. PK exposure similarity between loncastuximab tesirine total antibody and PBD-conjugated antibody indicated good stability in serum. Generally, exposure (area under the concentration-time curve [AUC] and maximum observed concentration [Cmax]) to loncastuximab tesirine was dose related and higher in cycle 2 than in cycle 1. As may be expected because of differing CD19 levels between patients, there was substantial variability in PK exposure and PK parameters assessed for PBD-conjugated antibody, total antibody, and SG3199. At 150 μg/kg, the mean half-life of PBD-conjugated antibody increased from 4.46 days in cycle 1 to 9.77 days in cycle 2, indicating likely moderate accumulation with multiple every-3-week treatment cycles. As expected, accumulation by cycle 2 for patients on an every-6-week dosing regimen was lower than that of those on every-3-week dosing: mean accumulations of 1.22 and 1.33 for PBD-conjugated antibody and total antibody on every-6-week regimens compared with 1.72 and 1.74 on every-3-week regimens, respectively.
Exposure-response modeling of the relationship between PBD-conjugated antibody exposure and TEAEs for 139 patients with available data (supplemental Table 6; supplemental Equation 1) showed a higher probability of grade ≥3 edema and liver enzyme abnormalities in the 200 μg/kg compared with 150 μg/kg dose cohort, with smaller comparative differences in probability of these events seen with 120 μg/kg compared with 150 μg/kg.
Immunogenicity
Of 183 patients tested for ADAs, 5 exhibited confirmed-positive ADAs predose, with low log2 titers (≤3), and 1 exhibited confirmed-positive ADAs postdose, with very low log2 titers (<1), indicating ADAs were not induced by loncastuximab tesirine.
Exploratory analysis
CD19+ tumor cells in tumor tissue ranged from 0% to 99%. No correlation was observed for CD19 expression in tumor tissue with PK exposure or with clinical response to treatment (supplemental Figure 1). From limited data on peripheral WBC changes (only US patients with ≥2 measurements), preliminary analysis suggested that CD19+ B cells were reduced compared with other cells upon treatment with loncastuximab tesirine. Baseline median numbers of peripheral CD19+ B cells per μL in serum were 58.0, 22.0, and 5.0 cells per µL for 120, 150, and 200 µg/kg doses, respectively. In 13 patients available for analysis who received doses of loncastuximab tesirine >90 μg/kg, median number of peripheral CD19+ B cells per μL in serum was reduced by ∼100% 7 days after the second dose, and the reduction was sustained until end of treatment. The percentage of peripheral CD19+ B cells significantly correlated with Cmax and AUC for PBD-conjugated antibody (Figure 4A-B). However, no relationship between CD19+ B-cell count and clinical response was observed.
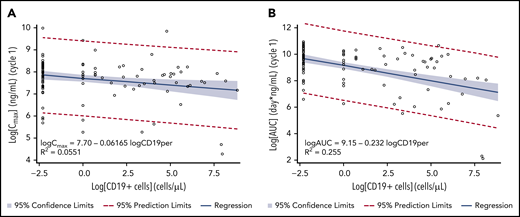
Correlation between Cmax and AUC of loncastuximab tesirine–conjugated antibody during cycle 1 and baseline peripheral CD19+ B cells. (A) Cmax. (B) AUC. Linear regression models were used with natural log of baseline peripheral CD19+ B-cell values as the independent variable and natural log of Cmax or AUC as the dependent variable. Zero baseline CD19+ cells was set to 0.1 cells per μL and LN(0.1) = −2.30. The estimated slope (standard error) was −0.0617 (0.0239; 95% CI, −0.109 to −0.0143) for Cmax and −0.232 (0.0370; 95% CI, −0.305 to −0.158) for AUC.
Correlation between Cmax and AUC of loncastuximab tesirine–conjugated antibody during cycle 1 and baseline peripheral CD19+ B cells. (A) Cmax. (B) AUC. Linear regression models were used with natural log of baseline peripheral CD19+ B-cell values as the independent variable and natural log of Cmax or AUC as the dependent variable. Zero baseline CD19+ cells was set to 0.1 cells per μL and LN(0.1) = −2.30. The estimated slope (standard error) was −0.0617 (0.0239; 95% CI, −0.109 to −0.0143) for Cmax and −0.232 (0.0370; 95% CI, −0.305 to −0.158) for AUC.
Discussion
In this phase 1 study of the ADC loncastuximab tesirine in patients with R/R B-NHL, a dosing regimen associated with reduction of tumor burden and an acceptable safety profile was established for further study. Four patients had DLTs (all hematologic), including 2 who received loncastuximab tesirine at 200 μg/kg; the MTD was not reached. TEAEs of hematologic abnormalities, peripheral edema, and liver test abnormalities were more common in the 200 μg/kg group than in lower-dose groups. The safety profile of loncastuximab tesirine was consistent with that previously reported for part 1,17 with no additional safety concerns during part 2. Toxicities were generally reversible and manageable in most patients with dose delays. Toxicities considered likely related to the PBD warhead were common, including edema and effusions, rash, and liver enzyme elevations.20,21 Incidences of edema and effusion were reduced in part 2 after introduction of dexamethasone premedication, and this approach is being employed to mitigate PBD-related toxicities in additional studies of loncastuximab tesirine, together with management with spironolactone. More stringent recommendations on sun exposure are intended to reduce rash.
Tumor burden was assessed, and tumor was found to be responsive to loncastuximab tesirine, with durable responses seen in a proportion of patients with DLBCL, MCL, and FL. Response rates with loncastuximab tesirine were generally lower in subgroups previously reported to have poorer prognosis, such as bulky disease, double-hit disease, and refractory DLBCL.2,8,22-25 However, a substantial proportion of patients with high-risk features had encouraging responses to loncastuximab tesirine, and characteristics of responders are being further elucidated in phase 2 studies, including analyses of response in activated B-cell vs germinal center B-cell subtypes, for which there was insufficient information for analysis in this phase 1 study.
A large proportion of the study population had R/R DLBCL, which is likely reflective of the unmet need for therapies in patients resulting from a lack of approved therapies at time of recruitment, together with higher incidence of this subtype. A number of treatments for patients with R/R DLBCL have been approved in recent years, including CAR T-cell therapies, polatuzumab vedotin-piiq with bendamustine and rituximab, selinexor, and tafasitamab-cxix with lenalidomide; however, an unmet need remains, because these therapies have substantial toxicities, and many patients do not have a durable response.26-29
Exposure to loncastuximab tesirine increased with dose. The similarity of conjugated and total antibody moieties in serum demonstrated good stability that could minimize systemic nonspecific toxicities that can occur with more labile ADCs.
Characterizing the effects of WBC level on drug exposure provides a fundamental understanding of drug action, because B cells bear the cognate target of loncastuximab tesirine. Both Cmax and AUC for PBD-conjugated antibody were significantly correlated with baseline CD19 expression. These significant relationships may explain, in part, the marked variability of PK exposure because of differences in baseline CD19 expression between patients. Exploratory analyses of target presence and response suggested target-mediated disposition could contribute to higher clearance, especially at the lower doses tested. The time-dependent component of clearance, thought to be related to clearance of CD19-expressing cells, was abrogated or eliminated by 5 cycles (∼15 weeks). Notably, there was no relationship between CD19 expression and clinical response to loncastuximab tesirine. Similar results have been reported for CD19- and CD30-directed therapies (tisagenlecleucel and brentuximab vedotin)30,31 in patients with B-NHL, with no correlation observed between target expression in tumor cells and clinical response. Consequently, determining the percentage of CD19+ tumor cells in tumor types known to express CD19 may have limited prognostic value.
Based on cumulative safety, PK, and efficacy data, the recommended dose of loncastuximab tesirine for phase 2 is 150 µg/kg every 3 weeks for 2 doses followed by 75 µg/kg every 3 weeks for subsequent doses. The 150 µg/kg dose was selected as a dose with encouraging responses but lower frequency of AEs than observed at the 200 µg/kg dose. Exposure-response modeling also demonstrated a higher probability of grade ≥3 edema and liver enzyme abnormalities in the 200 μg/kg compared with 150 μg/kg dose cohort. Smaller comparative differences in predicted probability were apparent between the 120 and 150 μg/kg doses. Moderate accumulation of loncastuximab tesirine together with frequent dose delays and 50% dose reductions after prolonged delays required during this study supported a strategy of planned dose reduction of 50% after 2 cycles to mitigate onset of late-developing and difficult-to-manage toxicities, such as edema, which generally developed after ≥2 cycles. Selection of this dosing regimen was further supported by the rapid onset of response (median, 2 cycles), and it is expected to optimize the frequency of objective response while reducing the need for dose delay or further dose reduction.
As demonstrated in this study, loncastuximab tesirine has substantial single-agent antitumor activity and is a promising off-the-shelf treatment option with outpatient administration for patients for whom multiple lines of therapy have failed, including patients unsuitable for HCT or CAR T-cell therapy or for whom such therapies have failed or as a bridge to such treatments. Notably, favorable outcomes have been reported in patients with R/R DLBCL treated with CAR T-cell therapy after previous loncastuximab tesirine treatment.32 As such, loncastuximab tesirine is being further investigated as monotherapy and in combination with other therapies (registered at www.clinicaltrials.gov as #NCT03684694, #NCT04384484, and #NCT03589469).
Study design is available at clinicaltrials.gov: NCT02669017. For original data, contact clinical.trials@adctherapeutics.com.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
Editorial assistance was provided by Becky Salisbury and Louise Gildea at Fishawack Communications, Ltd.
This study was supported by ADC Therapeutics, which also funded the Fishawack Communications, Ltd, editorial assistance. K.M.A. is supported by the University College London (UCL)/UCL Hospitals Biomedical Research Unit.
All authors meet the International Committee of Medical Journal Editors criteria for authorship and take responsibility for the integrity of the work as a whole. They also had full access to the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Authorship
Contribution: M.H., J.R., C.C.-S., P.F.C., E.R., O.A.O., K.M.A., W.T., M.S., L.T.H., and B.S.K. were involved in designing the study and data acquisition; J.M.F., D.U., J.B., and Y.Q. were involved in designing the study; K.H. and L.W. were involved in collecting and assembling the study data; and all authors were involved in data analysis and interpretation, contributed to the writing and reviewing of the manuscript, and have given final approval of the version to be published.
Conflict-of-Interest disclosure: M.H. has received research support from Takeda Pharmaceutical Company, Spectrum Pharmaceuticals, and Astellas Pharma; has served as a consultant for Janssen R&D, Incyte Corporation, ADC Therapeutics, Celgene Corporation, Pharmacyclics, Omeros, AbGenomics, Verastem, and TeneoBio; and has participated in speakers’ bureau for Sanofi Genzyme and AstraZeneca. J.R. has acted in a consultant/advisory role for Takeda, ADC Therapeutics, Bristol-Myers Squibb, Novartis, and Kite Pharma; has been a speaker for Takeda, ADC Therapeutics, and Seattle Genetics; has received research funding from Takeda; has provided expert testimony for Takeda and ADC Therapeutics; and holds stocks/shares in AstraZeneca and GlaxoSmithKline (spouse). C.C.-S. has received research support from ADC Therapeutics and Rhizen Pharmaceuticals; has served as a consultant or advisor for Servier, Novartis, Genenta Science srl, ADC Therapeutics, Roche, Boehringer Ingelheim, and Sanofi; and has received honoraria for speaker engagements from Bristol-Myers Squibb, Merck Sharp & Dohme, Janssen Oncology, and AstraZeneca. P.F.C., E.R., O.A.O., and M.S. have received research support from ADC Therapeutics. K.M.A. has received honoraria from or attended advisory boards for Celgene, Gilead, Takeda, Roche, and Beigene. W.T. has received research support from ADC Therapeutics and honoraria from Roche and Gilead. L.T.H. has received institutional research funding from Pharmacyclics, Genentech, Kite, and ADC Therapeutics and an honorarium from Kite. B.S.K. has received research support from ADC Therapeutics and acted as a consultant for Seattle Genetics and Genentech. J.M.F., D.U., L.W., J.B., K.H., and Y.Q. are employees of ADC Therapeutics with stock options.
Correspondence: Mehdi Hamadani, Division of Hematology and Oncology, Medical College of Wisconsin, 9200 W. Wisconsin Ave, Suite C5500, Milwaukee, WI 53226; e-mail: mhamadani@mcw.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal