

In this issue of Blood, 1 provide evidence in a prospective phase 4, multicentric, noncontrolled study that discontinuing eculizumab is safe in most patients with atypical hemolytic uremic syndrome (aHUS) once they achieve complete remission. Risk of relapse was <25% overall, but as high as 50% in patients with a rare variant in at least 1 complement gene.
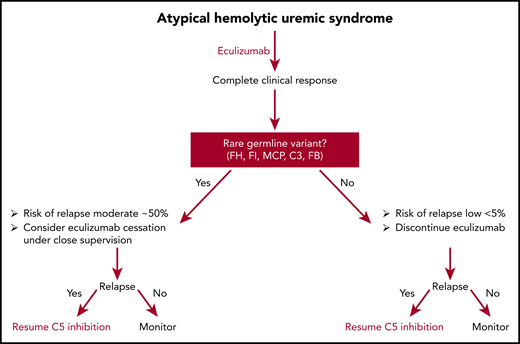
Potential algorithm for discontinuing eculizumab in patients who achieve complete remission.
Potential algorithm for discontinuing eculizumab in patients who achieve complete remission.
aHUS, commonly called complement-mediated hemolytic uremic syndrome (CM-HUS), is a thrombotic microangiopathy characterized by mechanical hemolysis, renal impairment, thrombocytopenia, and preserved ADAMTS13 function.2 CM-HUS results from complement-mediated damage to the microvascular endothelium. Kidneys are most vulnerable, but CM-HUS often affects multiple organs. Roughly 50% of patients have rare germline variants in complement genes or autoantibodies (less common) against complement regulatory proteins. Penetrance is variable because clinically significant disease requires a complement amplifying trigger such as infection, surgery, pregnancy, cancer, or autoimmunity.3 Most rare germline variants occur in genes that regulate the alternative pathway of complement. Complement factor H variants are most common (∼25%), followed by variants in membrane cofactor protein (∼10%) and complement factor I (5%-10%). A small percentage of patients (5%-10%) have autoantibodies against factor H; these patients often have homozygous deletion of complement factor–related genes. Activating mutations that prolong the half-life of C3 and complement factor B are also found in up to 5% of patients with CM-HUS. In 40% to 50% of patients, no rare variants or autoantibodies are evident, so absence of a germline variant does not exclude the diagnosis of CM-HUS. The diagnosis is established by the presence of a microangiopathic hemolytic anemia, thrombocytopenia, and renal insufficiency in a patient with an ADAMTS13 of >10% and a negative shiga toxin.
Therapeutic terminal complement blockade at the level of C5 (eculizumab or ravulizumab) changed the natural history of CM-HUS.4,5 Previously, more than 50% of patients died or acquired end-stage renal disease within 1 year of diagnosis; C5 inhibitors reduce this figure to less than 15%. Clinical improvement is observed within days to weeks after C5 inhibition, with most patients achieving a complete or near complete response after 3 to 6 months of therapy. It is unclear whether or not indefinite continuation of C5 inhibition is necessary for CM-HUS, a particularly important question, because C5 inhibition increases the patient’s risk for Neisseria meningitidis6 and costs more than $600 000 US dollars, annually. A few small retrospective series suggest that eculizumab discontinuation is safe and feasible in most patients but that relapse risk appears to be higher in carriers of rare complement gene variants7-9 The study by Fakhouri et al is the first prospective study of eculizumab discontinuation in patients with CM-HUS who achieved remission after eculizumab.
Fakhouri et al studied eculizumab discontinuation in 55 patients with CM-HUS who responded to C5 inhibition. Germline complement variants were present in 28 (51%) of patients. Remission was defined by a normal platelet count, the absence of hemolysis, and (1) a glomerular filtration rate of >60 mL per minute per 1.73 mm2 using the Schwartz formula in children and the Modification of Diet in Renal Disease formula in adults and a proteinuria/creatinuria ratio <0.05 g/mmol, or (2) stable kidney function for at least 6 months. During follow-up, 13 (23%) patients relapsed. In multivariate analysis, female sex and the presence of a rare germline complement variant were associated with an increased risk for relapse. Among the 13 patients with relapse, eculizumab was restarted, and 1l regained baseline renal function; 2 worsened, 1 of whom progressed to end-stage renal failure. The authors’ conclude that a strategy of eculizumab discontinuation in CM-HUS patients based on complement genetics is reasonable and safe.
This study increases our confidence about discontinuing eculizumab in most patients with CM-HUS who achieve remission. No patient without a germline complement mutation relapsed (1 relapsed patient was subsequently shown to have inherited ADAMTS13 deficiency); thus, it is fair to conclude that all patients with CM-HUS who achieve a complete remission after C5 inhibition deserve a trial off therapy, especially if the underlying complement amplifying condition is well controlled. Whether this holds for patients with factor H autoantibodies is unclear from the present study because there were too few patients. Another limitation of the study of Fakhouri et al is the composition of rare variants. Germline mutations in factor H are the most common mutations in CM-HUS, but not in this study. The most common mutation in this series was membrane cofactor protein (43%). Furthermore, the study included 9 patients (16%) who experienced more than 1 episode of CM-HUS (1 patient was included twice in the study) before study entry, potentially biasing the study and overestimating the relapse rate. Regardless, it is apparent that patients who carry rare germline variants are at greater risk for relapse, but even these patients have a relapse rate of 50% or less. Should we consider offering these patients a trial of eculizumab discontinuation under careful supervision? A case for such an approach could be made because virtually all patients recovered renal function after restarting terminal complement inhibition, but this requires more study (see figure). Finally, mean follow-up for this study was less than 18 months. Relapse from CM-HUS may occur decades later; moreover, we still do not understand all the late effects (risk for stroke, myocardial infarction, etc) that may be associated with CM-HUS even in the absence of overt relapse. Whether these late events are common and whether they can be prevented by complement inhibition requires future prospective trials.
Conflict-of-interest disclosure: R.A.B. reports grants and other from Alexion, outside the submitted work.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal