

Key Points
VPS45 is required for endocytic trafficking of G-CSFR and other cargos.
VPS45 is fundamental for early embryogenesis of mouse.

Abstract
Vacuolar protein sorting 45 homolog (VPS45), a member of the Sec1/Munc18 (SM) family, has been implicated in the regulation of endosomal trafficking. VPS45 deficiency in human patients results in congenital neutropenia, bone marrow fibrosis, and extramedullary renal hematopoiesis. Detailed mechanisms of the VPS45 function are unknown. Here, we show an essential role of mammalian VPS45 in maintaining the intracellular organization of endolysosomal vesicles and promoting recycling of cell-surface receptors. Loss of VPS45 causes defective Rab5-to-Rab7 conversion resulting in trapping of cargos in early endosomes and impaired delivery to lysosomes. In this context, we demonstrate aberrant trafficking of the granulocyte colony-stimulating factor receptor in the absence of VPS45. Furthermore, we find that lack of VPS45 in mice is not compatible with embryonic development. Thus, we identify mammalian VPS45 as a critical regulator of trafficking through the endosomal system and early embryogenesis of mice.
Introduction
Patients with loss-of-function variants in VPS45 present with infections due to severe neutropenia, progressive bone marrow fibrosis, and nephromegaly secondary to extramedullary hematopoiesis.1,2 Vacuolar protein sorting 45 homolog (VPS45) deficiency is further characterized by an impaired migration and increased apoptosis of neutrophil granulocytes. Notably, patients are refractory to the treatment with granulocyte colony-stimulating factor (G-CSF), even in high doses.1,2
VPS45 is involved in the endocytic pathway, which controls numerous cellular functions3-5 and is composed of various specialized compartments that are interconnected by vesicle flux.6,7 The first step of the endocytic pathway is the uptake of cargo material into preexisting early endosomes.8,9 Early endosomes function as sorting platforms, from which cargo is either recycled or destined for degradation.10,11
The endosomal system is highly dynamic and cargo transport along the endocytic route requires tight coordination. Rab GTPases and their effectors are critical regulators of the transport machinery.12-14 The small GTPase Rab5 governs vesicle fusion and intracellular organization of early endosomes,15-17 whereas the GTPase Rab7 controls late endocytic traffic.18 By gradually replacing Rab5 by Rab7, early endosomes mature into late endosomes.19,20 Upon fusion of late endosomes with lysosomes, cargo is proteolytically degraded in the lysosomal compartment. Conversely, recycling of cargo from Rab5-positive early endosomes to the cell surface is regulated by the GTPases Rab4 and Rab11, which characterize the fast and the slow recycling pathways, respectively.21-23 Because of its function as a dual Rab5 and Rab4 effector, Rabenosyn-5 plays a key role in the endocytic pathway.24,25 Besides Rab GTPases, Sec1/Munc18 (SM) proteins are essential to execute membrane fusion events by binding soluble N-ethylmaleimide-sensitive-factor attachment receptors.26 The evolutionarily conserved VPS45 protein is a member of the SM family and plays a crucial role in vesicle-mediated transport through the endosomal network. Mammalian VPS45 directly binds to Rabenosyn-525 as well as to the soluble N-ethylmaleimide-sensitive-factor attachment receptors protein Syntaxin 16,27 and the interaction is conserved in species, including Saccharomyces cerevisiae, Drosophila sp, and Caenorhabditis elegans.28-31 Studies in yeast suggest a role of Vps45p in the transport of vesicles from the Golgi apparatus to the prevacuolar compartment.32-34 Moreover, a function in cargo internalization and endosome formation has been ascribed to Vps45 in Drosophila and C elegans,29,30 and human VPS45 has been implicated in the recycling of β1 integrin.35
The precise function of the SM protein VPS45 in endosomal trafficking and its significance for mammalian development are not yet understood. In this study, we investigate the role of VPS45 in the endocytic pathway in vitro and in vivo.
Methods
Further details regarding the methods used are provided in the supplemental Methods, available on the Blood Web site.
Live cell imaging of endosome maturation
HeLa cells expressing mCherry-Rab5 and mCerulean-Rab7 were cultured in glass-bottomed dishes (ibidi; 35 mm number 1.5H) and incubated with fluorescent microspheres (FluoSpheres, carboxylate-modified microspheres, 0.02 µm, dark red fluorescent; Thermo Fisher Scientific) for 10 minutes at 37°C to allow uptake. Plates were washed twice and imaged in Dulbecco’s modified Eagle medium on a microscope stage at 37°C, 5% CO2 in a humidified atmosphere using a Zeiss LSM 800 confocal microscope using the Axio Observer.Z1/7 and a Plan-Apochromat 63×/1.4 oil immersion objective. Images were acquired every 12 seconds up to 90 minutes using bidirectional scanning and 1× line averaging.
Uptake and processing of cargo proteins
HeLa cells were incubated with either 32.5 µg/mL Alexa Fluor 488–conjugated ovalbumin (OVA-488; Thermo Fisher Scientific), 65 µg/mL of DQ ovalbumin (DQ OVA; Thermo Fisher Scientific), 65 µg/mL DQ Green bovine serum albumin (BSA; Thermo Fisher Scientific), or 65 µg/mL pHrodo Green dextran (Thermo Fisher Scientific) for various periods at 37°C. Cargo uptake and processing were stopped by washing the cells twice with ice-cold phosphate-buffered saline (PBS). The rate of cargo uptake and processing was determined by flow cytometry as a change in mean fluorescence intensity (MFI) from baseline (fluorescence at 4°C). When indicated, cells were treated with 25 µM chloroquine (CQ; Sigma-Aldrich). Alternatively, cells were processed for confocal microscopy.
EGF receptor trafficking assay
HeLa cells were serum-starved prior to stimulation with 100 ng/mL human epidermal growth factor (EGF; Thermo Fisher Scientific) for various time periods. Cells were washed twice in PBS and processed for immunoblotting or confocal microscopy.
G-CSFR trafficking assay
HeLa cells were transduced with granulocyte colony-stimulating factor receptor (G-CSFR)–green fluorescent protein fusion constructs and sorted to achieve equal expression levels. Cells were serum-starved, stimulated with 100 ng/mL recombinant human G-CSF (PeproTech) for various time periods, washed twice in PBS, fixed, and processed for confocal microscopy.
Results
VPS45 controls the intracellular organization of endolysosomal vesicles
To examine the function of human VPS45 in the endocytic system, we first generated clonal VPS45 knockout (KO) HeLa and PLB-985 cells using CRISPR/Cas9-mediated gene editing by targeting exon 7 of VPS45 (supplemental Figure 1A). Loss of VPS45 expression resulted in a markedly reduced abundance of its interacting proteins Rabenosyn-5 and Syntaxin 16 in both cell lines (Figure 1A; supplemental Figure 1B), demonstrating that VPS45 impacts on the expression of its interaction partners.
![VPS45 controls the intracellular organization of endolysosomal vesicles. (A) Immunoblot analysis of expression levels of VPS45 binding partners Rabenosyn-5 and Syntaxin 16 in control and VPS45 KO HeLa clones (n = 3 different clones per genotype). One representative experiment of 2 independent experiments is shown. (B) Immunofluorescence microscopy analysis of control and VPS45 KO HeLa clones to visualize early endosomes (EEA1), late endosomes (LAMP2), lysosomes (LAMP1), and the nucleus (4′,6-diamidino-2-phenylindole [DAPI]). One representative experiment of 3 independent experiments is shown. Scale bars, 10 µm. (C) Immunoblot analysis of expression levels of EEA1, LAMP2, and LAMP1 in control and VPS45 KO HeLa clones (n = 3 different clones per genotype). One representative experiment of 2 independent experiments is shown. ACTB, β-actin; Ctrl, control.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/14/10.1182_blood.2020006871/1/m_bloodbld2020006871f1.png?Expires=1765932712&Signature=NZtruHq88OGBeNH~Gr5s36Aw1D7q8LzNRRcrzGQa72ZGHyE64yRMWy8R2uzqHtTyke3nnJQaaj-GuwTdK5SqqjUZnEOuC4LOywTOVf9wPDH4~Rsd2kBe~CDCMviVAjjxSB0tAIT4nOOYtb4l0NlVxAsNcHAlImTg6DAIxqbIJds0jGxx0EIIY9hKtJR7CcG4hUshUXK3J5HJxCj6CiyAVEkGtuAaYUtMM-1sJV5iaVU~qYpa6tKasqdpoveI9AVeZ8XdjX9SHvpfJVpis68qQMHU-EEO1Zq4VzD6TseqZRutS3O7pbam7PX4mcBwD19n4hsO8B656jS~TJCS9RREGw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
VPS45 controls the intracellular organization of endolysosomal vesicles. (A) Immunoblot analysis of expression levels of VPS45 binding partners Rabenosyn-5 and Syntaxin 16 in control and VPS45 KO HeLa clones (n = 3 different clones per genotype). One representative experiment of 2 independent experiments is shown. (B) Immunofluorescence microscopy analysis of control and VPS45 KO HeLa clones to visualize early endosomes (EEA1), late endosomes (LAMP2), lysosomes (LAMP1), and the nucleus (4′,6-diamidino-2-phenylindole [DAPI]). One representative experiment of 3 independent experiments is shown. Scale bars, 10 µm. (C) Immunoblot analysis of expression levels of EEA1, LAMP2, and LAMP1 in control and VPS45 KO HeLa clones (n = 3 different clones per genotype). One representative experiment of 2 independent experiments is shown. ACTB, β-actin; Ctrl, control.
VPS45 controls the intracellular organization of endolysosomal vesicles. (A) Immunoblot analysis of expression levels of VPS45 binding partners Rabenosyn-5 and Syntaxin 16 in control and VPS45 KO HeLa clones (n = 3 different clones per genotype). One representative experiment of 2 independent experiments is shown. (B) Immunofluorescence microscopy analysis of control and VPS45 KO HeLa clones to visualize early endosomes (EEA1), late endosomes (LAMP2), lysosomes (LAMP1), and the nucleus (4′,6-diamidino-2-phenylindole [DAPI]). One representative experiment of 3 independent experiments is shown. Scale bars, 10 µm. (C) Immunoblot analysis of expression levels of EEA1, LAMP2, and LAMP1 in control and VPS45 KO HeLa clones (n = 3 different clones per genotype). One representative experiment of 2 independent experiments is shown. ACTB, β-actin; Ctrl, control.
Next, we studied the morphology and distribution of various types of intracellular vesicles. In striking contrast to control cells, in which the early endosomal marker EEA1 revealed a typical punctate distribution throughout the cytoplasm, EEA1-positive early endosomes were aggregated in VPS45 KO cells. Similarly, late endosomes expressing LAMP2 showed an aberrant morphology with clustered and enlarged vesicles. In addition, VPS45 deficiency resulted in an accumulation of LAMP1-positive lysosomes (Figure 1B). Despite the clustering of endosomal and lysosomal vesicles, the overall expression levels of EEA1, LAMP2, and LAMP1 were not altered in VPS45 KO cells (Figure 1C). These results document an essential role of VPS45 in maintaining the intracellular organization of endolysosomal vesicles.
Cargo recycling but not internalization and mitochondrial function depend on VPS45
To examine whether VPS45 deficiency also impairs cargo transport through these compartments, we tested the internalization, recycling, and degradation of various proteins. Both control and VPS45 KO cells were capable of endocytose fluorescent cargo at comparable rates over time, indicating that VPS45 is dispensable for cargo internalization (Figure 2A). In addition, the absence of VPS45 did not impact on mitochondrial function (supplemental Figure 2A-B).

VPS45 is dispensable for internalization but is required for efficient transferrin recycling. (A) Flow cytometric analysis of cargo uptake. Control and VPS45 KO HeLa clones were incubated with 32.5 µg/mL fluorescent OVA at 37°C for indicated periods, and the rate of cargo uptake was analyzed as a change in MFI from baseline (fluorescence at 4°C). Data are pooled from 2 independent experiments. Error bars indicate mean ± standard deviation (SD). Statistical analysis of significance using 2-way analysis of variance (ANOVA) followed by Tukey multiple comparisons test revealed no significant difference in OVA uptake between control and VPS45 KO HeLa clones. (B) Flow cytometric analysis of transferrin recycling in control and VPS45 KO HeLa clones. Cells were incubated with 25 µg/mL of fluorescent transferrin for 30 minutes at 37°C, followed by a chase for the indicated periods and removal of surface-bound transferrin. Representative histograms of 1 control clone and 1 VPS45 KO clone are shown. The chart shows cell-associated transferrin as the percentage of initial transferrin present at time t = 0, and pooled data from 4 independent experiments are shown. Error bars indicate mean ± SD. Statistical analysis was performed using 2-way ANOVA followed by Tukey multiple comparisons test. ns, not significant. ****P < .0001.
VPS45 is dispensable for internalization but is required for efficient transferrin recycling. (A) Flow cytometric analysis of cargo uptake. Control and VPS45 KO HeLa clones were incubated with 32.5 µg/mL fluorescent OVA at 37°C for indicated periods, and the rate of cargo uptake was analyzed as a change in MFI from baseline (fluorescence at 4°C). Data are pooled from 2 independent experiments. Error bars indicate mean ± standard deviation (SD). Statistical analysis of significance using 2-way analysis of variance (ANOVA) followed by Tukey multiple comparisons test revealed no significant difference in OVA uptake between control and VPS45 KO HeLa clones. (B) Flow cytometric analysis of transferrin recycling in control and VPS45 KO HeLa clones. Cells were incubated with 25 µg/mL of fluorescent transferrin for 30 minutes at 37°C, followed by a chase for the indicated periods and removal of surface-bound transferrin. Representative histograms of 1 control clone and 1 VPS45 KO clone are shown. The chart shows cell-associated transferrin as the percentage of initial transferrin present at time t = 0, and pooled data from 4 independent experiments are shown. Error bars indicate mean ± SD. Statistical analysis was performed using 2-way ANOVA followed by Tukey multiple comparisons test. ns, not significant. ****P < .0001.
Because the inactivation of VPS45 results in the reduced expression of the dual Rab4 and Rab5 effector Rabenosyn-5, we hypothesized that both Rab4- and Rab5-controlled recycling and degradative pathways might be affected in the absence of VPS45. The transferrin receptor and its ligand transferrin are widely used to study the recycling pathway. Control cells efficiently recycled transferrin, whereas VPS45 deficiency caused a delay in fast transferrin recycling (Figure 2B). An intracellular accumulation of transferrin after 10 and 30 minutes of recycling was also observed in VPS45 KO cells compared with control cells by confocal microscopy (supplemental Figure 2C). Endogenous expression levels of Rab4 and Rab11 were not affected by the depletion of VPS45 (supplemental Figure 2D). Thus, inactivation of VPS45 delays recycling of transferrin through the early endosomal sorting station.
VPS45 depletion disrupts cargo degradation and delays maturation of cathepsin D
To further evaluate whether the degradation pathway is affected in the absence of VPS45, we tested the transport of cargo destined for lysosomal processing using 3 distinct compounds. DQ OVA and DQ Green BSA exhibit fluorescence upon proteolytic processing in low pH compartments, whereas pHrodo Green dextran is pH sensitive and generates increasing amounts of fluorescent peptides with decreasing pH. Confocal microscopy analysis revealed efficient processing of DQ OVA in LAMP2-positive late endosomes in control cells after 90 minutes of incubation, whereas markedly less DQ OVA punctae were observed in VPS45 KO cells (Figure 3A). These results are in line with the reduced degradative capacity of VPS45 KO cells measured by flow cytometry (Figure 3B-D). Because VPS45 KO cells were capable of efficient compound uptake (Figure 2A), it is likely that postendocytic steps in the degradative pathway are affected. To further test the hypothesis of impaired fusion of endolysosomal vesicles, cells were additionally treated with CQ, a lysosomotropic component that raises the lysosomal pH and affects fusion activity.36-38 CQ treatment reduced DQ OVA fluorescence in control cells to the level of VPS45 KO cells, whereas it did not affect cargo degradation by VPS45 KO cells (Figure 3B). These results indicate a decreased degradative capacity in the absence of VPS45, consistent with a defective fusion of endolysosomal vesicles.

VPS45 KO cells fail to efficiently process soluble cargos and show a delayed maturation of cathepsin D. Control and VPS45 KO HeLa clones were incubated with various fluorescent substrates for indicated time periods (A-B: DQ OVA; C: DQ Green BSA; D: pHrodo Green Dextran). (A) Representative confocal microscopy images of DQ OVA processing in control and VPS45 KO HeLa clones are shown. After incubation with DQ OVA for 90 minutes, cells were fixed and stained for EEA1 and LAMP2. DAPI was used to visualize the nucleus. Enlarged images represent magnified views of boxed areas (8 µm × 8 µm). One representative experiment of 2 independent experiments is shown. Scale bars, 10 µm. (B-D) The rate of substrate processing was determined by flow cytometry as a change in MFI from baseline (fluorescence at 4°C). (B) Control and VPS45 KO cells were additionally treated with 25 µM CQ. Data are pooled from 4 independent experiments. (C) Data are pooled from 3 independent experiments. (D) Data are pooled from 4 independent experiments. (B-D) Error bars indicate mean ± SD. Statistical analysis was performed using 2-way ANOVA followed by Tukey or Sidak multiple comparisons test. *P < .05; ***P < 0,001; ****P < .0001. (E) Expression levels of immature (50 kDa), intermediate (46 kDa), and mature (28 kDa) cathepsin D were analyzed in control and VPS45 KO HeLa clones (n = 3 different clones per genotype) by immunoblotting. One representative experiment of 3 independent experiments is shown.
VPS45 KO cells fail to efficiently process soluble cargos and show a delayed maturation of cathepsin D. Control and VPS45 KO HeLa clones were incubated with various fluorescent substrates for indicated time periods (A-B: DQ OVA; C: DQ Green BSA; D: pHrodo Green Dextran). (A) Representative confocal microscopy images of DQ OVA processing in control and VPS45 KO HeLa clones are shown. After incubation with DQ OVA for 90 minutes, cells were fixed and stained for EEA1 and LAMP2. DAPI was used to visualize the nucleus. Enlarged images represent magnified views of boxed areas (8 µm × 8 µm). One representative experiment of 2 independent experiments is shown. Scale bars, 10 µm. (B-D) The rate of substrate processing was determined by flow cytometry as a change in MFI from baseline (fluorescence at 4°C). (B) Control and VPS45 KO cells were additionally treated with 25 µM CQ. Data are pooled from 4 independent experiments. (C) Data are pooled from 3 independent experiments. (D) Data are pooled from 4 independent experiments. (B-D) Error bars indicate mean ± SD. Statistical analysis was performed using 2-way ANOVA followed by Tukey or Sidak multiple comparisons test. *P < .05; ***P < 0,001; ****P < .0001. (E) Expression levels of immature (50 kDa), intermediate (46 kDa), and mature (28 kDa) cathepsin D were analyzed in control and VPS45 KO HeLa clones (n = 3 different clones per genotype) by immunoblotting. One representative experiment of 3 independent experiments is shown.
Expression of the mature form of the protease cathepsin D can be used as an indicator of the lysosomal pH and proteolytic capacity, as the conversion of cathepsin D from the 53-kDa immature and 47-kDa intermediate forms to the 31-kDa mature form requires transport to late endosomes or lysosomes and an appropriate acidic milieu. Whereas mature forms of cathepsin D were virtually absent, immature forms accumulated in VPS45 KO cells (Figure 3E). These results are in line with the reduced protein degradation capacity and could reflect an inefficient delivery of proteases from the Golgi apparatus to endolysosomal vesicles in the absence of VPS45. Of note, the maturation of cathepsin D was further inhibited by incubation with CQ, indicating that lysosomal transport and biogenesis are not merely dependent on VPS45 (supplemental Figure 3).
EGFR is accumulated in early endosomes in the absence of VPS45
Next, we examined trafficking of epidermal growth factor receptor (EGFR), a model receptor that is used to study trafficking along the degradative pathway. In control cells, a substantial degradation of receptors was observed over time. In contrast, deletion of VPS45 prevented efficient degradation of EGFR (Figure 4A). To gain further insight into EGFR trafficking defects, we visualized the fate of EGFR. After 10 minutes of stimulation, the receptor was enriched in EEA1-positive early endosomes in both control and VPS45 KO cells, indicating an intact internalization. After 90 minutes, EGFR signals revealed reduced colocalization with EEA1-positive endosomes and an overall decrease in control cells, suggesting an efficient exit of the cargo from early endosomes. In contrast, EGFR remained captured in EEA1-positive vesicles in VPS45 KO cells over time (Figure 4B-C). These data suggest that VPS45 facilitates the fusion of early endosomal and lysosomal vesicles.
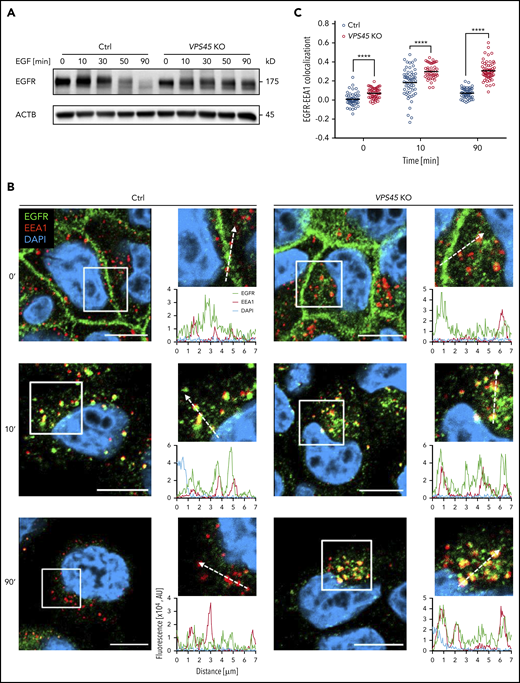
Loss of VPS45 causes accumulation of EGFR in early endosomes and inefficient degradation. (A) Serum-starved control and VPS45 KO HeLa clones were stimulated with 100 ng/mL EGF for indicated time periods, and EGFR levels were analyzed by immunoblotting. One representative experiment of 3 independent experiments is shown. (B) Cells were treated as in panel A, fixed, and costained with anti-EGFR and anti-EEA1 antibodies. Nuclei were visualized using DAPI. Enlarged images represent magnified views of boxed areas (10 × 10 µm), and profile blots along the dashed arrow (7 µm) are shown. One representative experiment of 3 independent experiments is shown. Scale bars, 10 µm. (C) Quantification of EGFR-EEA1 colocalization of data shown in panel B. Pearson correlation coefficient is plotted on the y-axis. For each genotype, ≥40 cells were analyzed. Horizontal lines indicate the means, and each symbol represents an individual cell. Statistical analysis was performed using 2-way ANOVA with Tukey multiple comparisons test. ****P < .0001.
Loss of VPS45 causes accumulation of EGFR in early endosomes and inefficient degradation. (A) Serum-starved control and VPS45 KO HeLa clones were stimulated with 100 ng/mL EGF for indicated time periods, and EGFR levels were analyzed by immunoblotting. One representative experiment of 3 independent experiments is shown. (B) Cells were treated as in panel A, fixed, and costained with anti-EGFR and anti-EEA1 antibodies. Nuclei were visualized using DAPI. Enlarged images represent magnified views of boxed areas (10 × 10 µm), and profile blots along the dashed arrow (7 µm) are shown. One representative experiment of 3 independent experiments is shown. Scale bars, 10 µm. (C) Quantification of EGFR-EEA1 colocalization of data shown in panel B. Pearson correlation coefficient is plotted on the y-axis. For each genotype, ≥40 cells were analyzed. Horizontal lines indicate the means, and each symbol represents an individual cell. Statistical analysis was performed using 2-way ANOVA with Tukey multiple comparisons test. ****P < .0001.
Lack of VPS45 causes trapping of G-CSFR in early endosomes and hampered delivery to late endosomes
Patients with loss-of-function variants in VPS45 are neutropenic and refractory to G-CSF treatment.1,2 Our data demonstrate an impaired endolysosomal vesicle fusion in the absence of VPS45. Hence, we hypothesized that hyporesponsiveness to G-CSF is caused by impaired trafficking of G-CSFR. Upon ligand-binding, G-CSFR is internalized and transported to early endosomes, where most of the receptor-ligand complex is sorted for lysosomal degradation.39 After 30 minutes of stimulation, the G-CSFR/G-CSF complex reached EEA1-positive early endosomes in both control and VPS45 KO cells. Whereas G-CSFR colocalized with LAMP2-positive late endosomes after 30 and 90 minutes in control cells, trafficking to late endosomes was impaired in VPS45 KO cells, which showed a sustained accumulation of receptors in early endosomes instead (Figure 5A-B). Of note, receptor mistrafficking did not impact markedly on signaling, as G-CSFR- and GM-CSFR-mediated signaling seemed to be intact in the absence of VPS45 (supplemental Figure 4A-C). However, more studies are required to study the potential impact of G-CSFR mistrafficking on the refractoriness of VPS45-deficient patients to G-CSF therapy in depth.

VPS45 depletion results in prolonged association of G-CSFR with early endosomes and disrupted transport to late endosomes. (A) Serum-starved control and VPS45 KO HeLa clones stably expressing G-CSFR–green fluorescent protein fusion proteins were stimulated with 100 ng/mL G-CSF for 10 minutes, followed by a chase for the indicated periods. Stimulated cells were fixed and stained with anti-EEA1 and anti-LAMP2 antibodies. DAPI was used as a nuclear stain. Enlarged images represent magnified views of boxed areas (10 × 10 µm). One representative experiment of 2 independent experiments is shown. Scale bars, 10 µm. (B) Quantification of G-CSFR-EEA1 and G-CSFR-LAMP2 colocalization of data shown in panel A. Pearson correlation coefficient is plotted on the y-axis. For each genotype, ≥100 cells were analyzed. Horizontal lines indicate the means, and each symbol represents an individual cell. Statistical analysis was performed using 2-way ANOVA with Tukey multiple comparisons test. ****P < .0001.
VPS45 depletion results in prolonged association of G-CSFR with early endosomes and disrupted transport to late endosomes. (A) Serum-starved control and VPS45 KO HeLa clones stably expressing G-CSFR–green fluorescent protein fusion proteins were stimulated with 100 ng/mL G-CSF for 10 minutes, followed by a chase for the indicated periods. Stimulated cells were fixed and stained with anti-EEA1 and anti-LAMP2 antibodies. DAPI was used as a nuclear stain. Enlarged images represent magnified views of boxed areas (10 × 10 µm). One representative experiment of 2 independent experiments is shown. Scale bars, 10 µm. (B) Quantification of G-CSFR-EEA1 and G-CSFR-LAMP2 colocalization of data shown in panel A. Pearson correlation coefficient is plotted on the y-axis. For each genotype, ≥100 cells were analyzed. Horizontal lines indicate the means, and each symbol represents an individual cell. Statistical analysis was performed using 2-way ANOVA with Tukey multiple comparisons test. ****P < .0001.
VPS45 depletion results in defective endosome maturation
Endosome maturation ensures that cellular cargo targeted for degradation reaches the lysosomal compartment. This process is defined by the conversion of early endosomal Rab5 to late endosomal Rab7.19 We observed the arrest of internalized cargo proteins and receptors in early endosomes in the absence of VPS45, suggesting an impaired endosome maturation. Of note, endogenous levels of Rab5 and Rab7 were not affected in VPS45 KO cells (supplemental Figure 5). To further assess whether the switch of Rab5 to Rab7 is affected in VPS45 KO cells, the fate of fluorescent cargo was monitored by live cell imaging in control and VPS45 KO cells coexpressing mCherry-Rab5 and mCerulean-Rab7. Notably, the overall appearance of early and late endosomal vesicles differed between control and VPS45 KO cells. Ring-shaped structures for both Rab5- and Rab7-positive vesicles were only observed in control cells (Figure 6A). In VPS45 KO cells, vesicles were aggregated and disorganized, like vesicles marked by EEA1, LAMP2, and LAMP1 proteins (Figure 1B).
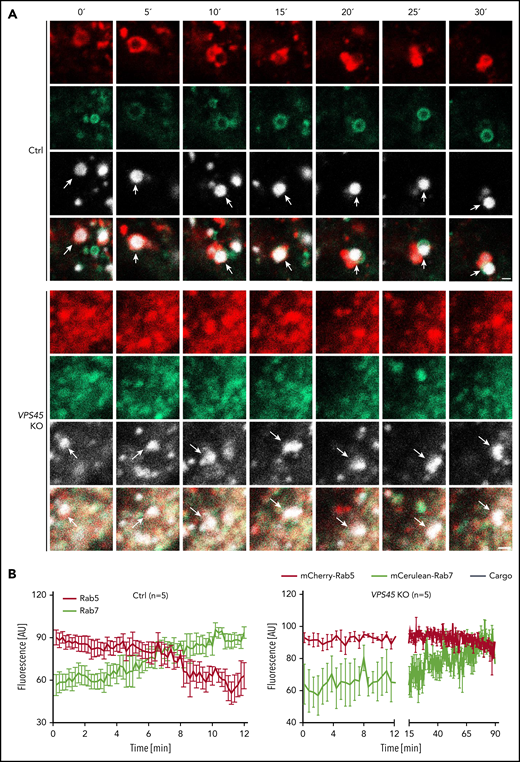
Loss of VPS45 leads to a defective early-to-late endosome maturation. Nonmodified control and VPS45 KO HeLa clones were transduced with recombinant lentiviral vectors to coexpress mCherry-Rab5 and mCerulean-Rab7. Dynamic changes of Rab5-positive and Rab7-positive vesicles were imaged during the uptake and transport of fluorescent cargo (microspheres-633). Arrows point to microspheres. (A) Representative images of cargo trafficking in single vesicles of control and VPS45 KO clones are shown. Scale bars, 1 µm. (B) The corresponding total fluorescence traces of Rab5 (red) and Rab7 (green) over time averaged from 5 independent experiments are shown. For visualization purposes, only every second frame is shown for the first 12 minutes in VPS45 KO clones. Error bars indicate mean ± standard error of the mean.
Loss of VPS45 leads to a defective early-to-late endosome maturation. Nonmodified control and VPS45 KO HeLa clones were transduced with recombinant lentiviral vectors to coexpress mCherry-Rab5 and mCerulean-Rab7. Dynamic changes of Rab5-positive and Rab7-positive vesicles were imaged during the uptake and transport of fluorescent cargo (microspheres-633). Arrows point to microspheres. (A) Representative images of cargo trafficking in single vesicles of control and VPS45 KO clones are shown. Scale bars, 1 µm. (B) The corresponding total fluorescence traces of Rab5 (red) and Rab7 (green) over time averaged from 5 independent experiments are shown. For visualization purposes, only every second frame is shown for the first 12 minutes in VPS45 KO clones. Error bars indicate mean ± standard error of the mean.
In control cells, a continuous decline in the early endosomal marker Rab5 and a gradual increase in the late endosomal marker Rab7 on cargo vesicles were observed over time. Eventually, Rab5 completely dissociated from the cargo while being replaced by Rab7, indicative of complete endosome maturation (Figure 6A-B; supplemental Video 1). In VPS45 KO cells, cargo was transported to Rab5-positive vesicles. Even though we observed that Rab7 appeared on Rab5-positive endosomes, Rab7 recruitment was slower in comparison with control cells. Importantly, in contrast to control cells, Rab5 did not disappear from the cargo vesicle in VPS45 KO cells, suggesting that endosome maturation was never fully completed (Figure 6A-B; supplemental Video 2). These data show that VPS45 is required for early-to-late endosome conversion and efficient cargo delivery to the degradative compartment.
Alleles carrying patient-associated VPS45 variants are hypomorphic
In contrast to the full VPS45 KO in engineered HeLa cells, patients with biallelic point variants in the VPS45 gene retain some residual protein expression. It is unclear to which degree variant VPS45 proteins are still functional. To further investigate the effects of the patient-associated Glu238Lys and Thr224Asn variants on the VPS45 proteins, we analyzed their subcellular localization. Confocal microscopy analysis revealed no difference in localization between control and variant VPS45 proteins (supplemental Figure 6A).
We next investigated whether these variants abolish the interaction of VPS45 with the known binding partners Rabenosyn-5 and Syntaxin 16. HEK293T cells were transiently transfected with C-terminally FLAG-tagged control or variant VPS45 plasmids. In these cells, we found that variant VPS45 was significantly less abundant than control VPS45 (supplemental Figure 6B). Copurification results confirmed interactions of control VPS45 with Rabenosyn-5 and Syntaxin 16, respectively. Moreover, variant VPS45 proteins were able to pull down both interaction partners, albeit to a lesser degree (supplemental Figure 6C). These results indicate that the interactions of variant VPS45 and binding partners are not completely abolished. Importantly, degradation rates of DQ OVA were restored by overexpressing either control or variant versions of VPS45 in KO cells (supplemental Figure 6D), suggesting that the alleles in VPS45-deficient patients are hypomorphic.
By introducing the patient-specific Thr224Asn variant into induced pluripotent stem cells (iPSCs), we further analyzed its effect on neutrophil development. During in vitro differentiation, the expression of stemness-related and granule genes as well as the percentage of CD33lowCD11b+ was comparable in control and VPS45 KI neutrophils (supplemental Figure 7A-C); however, significantly fewer live floating cells were counted in wells containing VPS45 KI colonies, indicating a reduced capacity to generate neutrophils (supplemental Figure 7D). This is in line with the observation of multiple apoptotic cells in these cells by confocal microscopy. In nonapoptotic cells, the cytoplasmic localization of neutrophil elastase, a protein frequently mutated in severe congenital neutropenia, was similar for both genotypes (supplemental Figure 7E). Of note, the generation of VPS45 KO iPSCs was not successful, indicating that a complete loss might be lethal.
Lack of VPS45 in mouse development is lethal at around E7.5
To further explore the biological relevance of VPS45 in a mammalian in vivo model, we generated a Vps45 KO mouse. Because all patient-associated variants maintain partial functionality, we hypothesized that a complete loss of Vps45 expression might result in embryonic lethality in mice. Therefore, we employed the KO-first strategy to generate the VPS45-deficient mouse model, as this strategy provides the flexibility to rapidly convert the null allele into a conditional allele40 (supplemental Figure 8A). One hundred forty-eight female and male pups from 24 litters of heterozygous breeding pairs were genotyped (Figure 7A). Of these pups, 37% (55 mice) were homozygous for the control allele and 63% (93 mice) were heterozygous for the Vps45 null allele. None of the pups was homozygous for the Vps45 null allele, indicating that the complete deletion of Vps45 is embryonic lethal for mice (Figure 7B).

VPS45 is essential for mouse embryogenesis at an early stage. (A) Representative polymerase chain reaction–based genotyping results of postnatal progeny derived from Vps45 heterozygous intercrosses. Primer sets 1 and 2 used to identify control and heterozygous (Het) mice are shown in supplemental Figure 5A. M, DNA marker. (B) Genotyping results of 148 postnatal pups derived from Vps45 heterozygous intercrosses. (C) A representative uterus derived from heterozygous intercrosses dissected at E13. Arrowheads show smaller implantation sites, indicating embryos that either died or were severely delayed in development. Scale bar, 5 mm. (D) Representative pictures of excised embryos derived from Vps45 heterozygous intercrosses at E13, E11, and E7.5. Scale bars, 5 mm. (E) PAS-stained cross sections of paraffin-embedded E7.5 embryos derived from Vps45 heterozygous intercrosses showing the embryonic ectoderm (EE), endoderm (EN), and mesoderm (ME). Scale bars, 1 mm.
VPS45 is essential for mouse embryogenesis at an early stage. (A) Representative polymerase chain reaction–based genotyping results of postnatal progeny derived from Vps45 heterozygous intercrosses. Primer sets 1 and 2 used to identify control and heterozygous (Het) mice are shown in supplemental Figure 5A. M, DNA marker. (B) Genotyping results of 148 postnatal pups derived from Vps45 heterozygous intercrosses. (C) A representative uterus derived from heterozygous intercrosses dissected at E13. Arrowheads show smaller implantation sites, indicating embryos that either died or were severely delayed in development. Scale bar, 5 mm. (D) Representative pictures of excised embryos derived from Vps45 heterozygous intercrosses at E13, E11, and E7.5. Scale bars, 5 mm. (E) PAS-stained cross sections of paraffin-embedded E7.5 embryos derived from Vps45 heterozygous intercrosses showing the embryonic ectoderm (EE), endoderm (EN), and mesoderm (ME). Scale bars, 1 mm.
To define the developmental stage at which Vps45 plays an essential role, embryos were analyzed at different days post coitum. Most of the embryos isolated at embryonic day 11 (E11) and E13 showed an age-appropriate development. However, some embryos were markedly smaller and did not develop properly (Figure 7C-D). Genotyping showed that these embryos were homozygous for the Vps45 null allele (supplemental Figure 8B). No visible differences in embryo size and shape were detected at E7.5 (Figure 7D). Histological analysis of E7.5 embryos revealed organized embryonic endodermal, mesodermal, and ectodermal structures, suggesting that those embryos are either homozygous for the control allele or heterozygous for the Vps45 null allele. In contrast, cell layers of suspected Vps45 KO embryos revealed an overall smaller cell mass and appeared as disorganized structures, making discrimination of germ layers impossible (Figure 7E). Overall, the formation of germ layers appeared to be impaired, pointing toward a defect in the process of gastrulation in the absence of VPS45.41
Because the global loss of VPS45 in mice resulted in embryonic lethality at around E7.5 and prevented us from studying the role of VPS45 for neutrophil differentiation, we generated 2 Vps45fl/fl;LysM-Cre or Vps45fl/fl;Vav-Cre conditional KO mouse lines to deplete VPS45 in the myeloid and hematopoietic compartments, respectively. However, VPS45 was inefficiently deleted in sorted neutrophils of Vps45fl/fl;LysM-Cre and bone marrow cells of Vps45fl/fl;Vav-Cre conditional KO mice and thus precluded further analysis of neutrophil differentiation (supplemental Figure 8C-D).
VPS45 depletion delays neutrophil differentiation ex vivo
To circumvent the obstacle of studying neutrophil differentiation in vivo, we opted to delete VPS45 ex vivo using Cre-expressing retroviral vectors. Cre-expressing myeloid CD11b+Gr1+ cells carrying homozygous floxed, heterozygous floxed, or homozygous control Vps45 alleles showed lowest, intermediate, and highest Vps45 messenger RNA levels, respectively (supplemental Figure 9A). Generally, the percentage of Cre-positive neutrophils declined over time, consistent with Cre-mediated cellular toxicity. Numbers of VPS45-depleted neutrophils significantly decreased at day 4 of coculture, and a further decline was observed at day 7 as compared with control cells (supplemental Figure 9B-C). Insufficient Vps45 expression may have led to increased cell death or a halt in differentiation. These results stress the significance of VPS45 for neutrophil differentiation.
Discussion
In this study, we report a novel role of VPS45 in regulating endocytic trafficking, particularly the fusion of early endosomes. Complete loss of VPS45 results in the perturbed intracellular organization of endolysosomal vesicles and cargo mistrafficking through the early endosomal compartment. We demonstrate that VPS45 deficiency causes aberrant trafficking of G-CSFR, which might be associated with the severe neutropenic phenotype of VPS45-deficient patients and their poor response to G-CSF therapy. Importantly, we find that lack of VPS45 is not compatible with life in the mouse.
Previous work has shown that a reduction of VPS45 expression due to either patient-associated variants or small interfering RNA–mediated knockdown results in decreased amounts of binding partners Rabenosyn-5 and Syntaxin 16.2,35 We here confirm that VPS45 regulates the expression levels of these proteins, as Rabenosyn-5 and Syntaxin 16 levels are reduced to an even higher extent in the complete absence of VPS45. The VPS45 protein may be critical to stabilize these binding partners and prevent degradation of the complex members via the proteasomal pathway.35 We assume that reduced amounts of the vesicle fusion machinery components VPS45, Rabenosyn-5, and Syntaxin 16 may impede efficient vesicle fusion, resulting in the accumulation of endolysosomal vesicles. This accords with studies in yeast and Drosophila, in which large aggregates of potential transport vesicles are found in Vps45 null cells,30,33 pointing toward an evolutionarily conserved function of VPS45 in fusion events of these cargo vesicles in the endosomal system.
Studies in Drosophila and C elegans describe a role of both VPS45 and Rabenosyn-5 in cargo internalization.29,30 In contrast, we find that VPS45 is dispensable for endocytosis in human cells, because neither clathrin-mediated endocytosis of transferrin, EGFR, or G-CSFR, nor clathrin-independent uptake of OVA and microspheres are affected in the absence of VPS45. Our results further contradict a study suggesting an impact of VPS45 on uptake and hence mitochondrial function in the pathogenic fungus Cryptococcus neoformans.40 Because we find that lack of VPS45 does not affect uptake in human cells, mitochondrial function remains intact. However, we find that both recycling and degradation pathways are perturbed in VPS45 KO cells.
The phenotype of VPS45 KO cells may in part be due to reduced Rabenosyn-5 abundance, known to orchestrate both recycling and degradation pathways. For example, Rabenosyn-5 controls transferrin recycling,42 presumably because of its function as an effector of Rab4, which in turn is known to promote the fast recycling pathway.22,24 Of note, small interfering RNA–mediated knockdown of Rabenosyn-5 in monkey cells resulted in mislocalization and impaired uptake of transferrin, pointing toward divergent roles of VPS45 and Rabenosyn-5 in the endocytic pathway.42 Rabenosyn-5 also interacts with EHD1, a protein involved in the slow recycling route.43,44 Thus, it is plausible that the absence of VPS45 and lower levels of Rabenosyn-5 resulting thereof lead to a delay in transferrin recycling. We also observe vesicle fusion defects at early endosomes, where Rabenosyn-5 is known to act as a Rab5 effector molecule.25 The switch from early endosomal Rab5 to late endosomal Rab7 is impaired, presumably because of the lack of Rabenosyn-5. Defective endosome maturation results in an accumulation of a variety of cargos in the early endosomal compartment and impaired transfer to the degradative compartment. Likewise, absent VPS45 might impact on the transport and delivery of cathepsin D to the low pH compartment, thus affecting the maturation of the lysosomal protease. In addition, Syntaxin 16 is reported to localize to the Golgi apparatus and to function in the retrograde transport to the TGN.45,46 Reduced expression of Syntaxin 16 could thus affect the mannose 6–dependent transport of the lysosomal protease cathepsin D as well.
Of note, VPS45 and Rabenosyn-5 deficiencies exhibit phenotypic similarities. VPS45-deficient patients suffer from severe neutropenia associated with myelofibrosis.1,2,47 Similarly, Rabenosyn-5–deficient patients present with syndromic myelofibrosis associated with neutropenia and bone marrow failure.48,49 VPS45-deficient patients do not respond to G-CSF treatment,1,2,47 a phenotype also observed for Rabenosyn-5–deficient patients.48 We find that EEA1-positive early endosomes are aggregated in the absence of VPS45 in HeLa cells. Similarly, early endosomes were enlarged and clustered in fibroblasts of Rabenosyn-5–deficient patients.48 Moreover, transferrin recycling is delayed in VPS45 KO HeLa cells, which is also the case for fibroblasts derived from Rabenosyn-5–deficient patients.48 The complete loss of either Rabenosyn-5 (phenotyping center, Wellcome Trust Sanger Institute) or VPS45 causes preweaning or early embryonic lethality in mice, respectively. Based on our characterization of the variant VPS45 proteins, we assume that they behave as hypomorphs. Overexpression of the variant constructs in VPS45 KO HeLa cells provides cells with sufficient amounts of partially functional variant VPS45 proteins to restore endosome maturation and cargo delivery to the degradative compartment. In contrast, residual VPS45 levels in patient cells may be too low to efficiently exert function in vesicle transport. At this point, we cannot fully explain to what extent the level of protein expression and the loss-of-function variant contribute to residual VPS45 activity.
In our study, we could confirm a critical role of mammalian VPS45 for neutrophil granulocyte differentiation. However, it remains elusive why neutrophils are particularly sensitive to disruption of VPS45-mediated vesicle trafficking. Based on our findings, VPS45 regulates recycling and degradation pathways crucial for multiple immune cells. We would thus expect a broader defect rather than neutropenia. On the other hand, variants in a variety of genes have been identified to affect endosome integrity and to cause severe neutropenia. Human defects in VPS13B,50 RAB27A,51 and LAMTOR252 highlight the necessity of proper vesicle trafficking pathways for neutrophil granulocytes. We thus propose that aberrant assembly and fusion of granules with phagolysosomes might be the underlying pathomechanism of VPS45 deficiency, leading to inefficient destruction of engulfed pathogens or dysregulated signaling of receptors.
Taken together, we here demonstrate that loss of VPS45 severely disrupts the intracellular organization of endolysosomal vesicles and interferes with efficient cargo recycling and degradation as well as the endosome maturation. VPS45 is critical for early embryonic development in the mouse. Thus, we provide mechanistic insights into a rare hematopoietic disorder characterized by bone marrow fibrosis and congenital neutropenia.
For original data, please e-mail the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by grants from the following institutions: the Deutsche Forschungsgemeinschaft (DFG) (SFB914 and Gottfried Wilhelm Leibniz Program), the Bundesministerium für Bildung und Forschung (BMBF) (German PID-NET), the Care-for-Rare Foundation, the Deutsches Zentrum für Infektionsforschung (DZIF), and the Elite Network of Bavaria (“i-Target” doctoral program).
Authorship
Contribution: L.F., N.Z., M.Ł., and C.K. designed the research; L.F. conducted experiments and analyzed the data; Y.M. generated genetically modified iPSCs; M.I.L. assisted in iPSCs differentiation; B.M. and Y.L. assisted in molecular cloning and CRISPR/Cas9-mediated gene editing; F.G. and W.W. performed mouse rederivation by in vitro fertilization and helped with the embryo analysis, M.D., M.R.S., and E.W. generated the KO mice; R.S. provided critical interpretation of the data; N.Z., M.Ł., and C.K. conceived the study design and supervised L.F.; L.F. wrote the manuscript; N.Z., M.Ł., and C.K. revised the manuscript; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Christoph Klein, Department of Pediatrics, Dr von Hauner Children’s Hospital, Ludwig-Maximilians-Universität München, Lindwurmstraße 4, 803377 Munich, Germany; e-mail: christoph.klein@med.uni-muenchen.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal