

Key Points
The recommended phase 2 dose of InO for pediatric patients with ALL was established at 1.8 mg/m2 per course.
Of the patients with multiple R/R ALL, 85% reached CR after 1 course of single-agent InO at the RP2D, 100% of whom had MRD negativity.

Abstract
This phase 1 study investigated the recommended phase 2 dose (RP2D) of inotuzumab ozogamicin (InO), a CD22-directed antibody-drug conjugate, in pediatric patients with multiple relapsed/refractory (R/R) CD22+ acute lymphoblastic leukemia (ALL). Patients (age ≥1 year or <18 years) received 3 doses of InO (days 1, 8, and 15) per course. Dose escalation was based on dose-limiting toxicities (DLTs) during course 1. Dose level 1 (DL1) was 1.4 mg/m2 (0.6, 0.4, 0.4 mg/m2) and DL2 was 1.8 mg/m2 (0.8, 0.5, 0.5 mg/m2). Secondary end points included safety, antileukemic activity, and pharmacokinetics. Twenty-five patients (23 evaluable for DLTs) were enrolled. In course 1, the first cohort had 1 of 6 (DL1) and 2 of 5 (DL2) patients who experienced DLTs; subsequent review considered DL2 DLTs to be non–dose-limiting. Dose was de-escalated to DL1 while awaiting protocol amendment to re-evaluate DL2 in a second cohort, in which 0 of 6 (DL1) and 1 of 6 (DL2) patients had a DLT. Twenty-three patients experienced grade 3 to 4 adverse events; hepatic sinusoidal obstruction syndrome was reported in 2 patients after subsequent chemotherapy. Overall response rate after course 1 was 80% (95% confidence interval [CI], 59% to 93%) (20 of 25 patients; DL1: 75% [95% CI, 43% to 95%], DL2: 85% [95% CI, 55% to 98%]). Of the responders, 84% (95% CI, 60% to 97%) achieved minimal residual disease (MRD)-negative complete response, and 12-month overall survival was 40% (95% CI, 25% to 66%). Nine patients received hematopoietic stem cell transplantation or chimeric antigen receptor T cells after InO. InO median maximum concentrations were comparable to simulated adult concentrations. InO was well tolerated, demonstrating antileukemic activity in heavily pretreated children with CD22+ R/R ALL. RP2D was established as 1.8 mg/m2 per course, as in adults. This trial was registered at https://www.clinicaltrialsregister.eu as EUDRA-CT 2016-000227-71.
Introduction
Acute lymphoblastic leukemia (ALL) is the most frequent malignant disease of childhood, with an estimated western European incidence of 40 cases per million per year in children age younger than 15 years.1 In the past 2 decades, significant improvement has been made in treating childhood ALL, which is now curable in >85% of children with multidrug front-line protocols.2-4 However, the prognosis for those children who relapse or who are refractory to conventional therapy (∼15%) remains poor.4-6 Only about 50% of children who have relapsed can be rescued with intensive chemotherapy, followed by allogeneic hematopoietic stem cell transplantation (HSCT) in high-risk cases.4 Therefore, new therapeutic approaches are urgently needed to overcome chemotherapy resistance and improve outcome.
Inotuzumab ozogamicin (InO) is an antibody-drug conjugate composed of a monoclonal CD22-directed antibody linked to calicheamicin,7 a potent cytotoxic antitumor antibiotic that causes cell death by inducing double-strand DNA breaks.8,9 CD22 is a B-cell adhesion molecule that is expressed on both normal and malignant B cells. It is expressed in ∼90% of patients with childhood B-cell precursor ALL (BCP-ALL).10 InO is approved for treating adults with CD22+ relapsed or refractory (R/R) BCP-ALL, starting at 1.8 mg/m2 per course (fractionated schedule).7,11-13 Results from an adult phase 3 study revealed superiority of InO given once per week over standard intensive chemotherapy, with significantly greater rates of complete response (CR), minimal residual disease (MRD) negativity, and number of patients proceeding to transplantation.9 Liver-related, treatment-emergent events, including hepatic sinusoidal obstruction syndrome (SOS), were the most frequent nonhematologic toxicities.9
Given the results of InO in adult ALL and the medical need in pediatric R/R ALL, investigation of InO in pediatric BCP-ALL is highly warranted. In vitro data suggested high sensitivity of childhood ALL cells to calicheamicin.14 Existing data on InO from a phase 2 study (5 children) and compassionate use programs in Europe-United States (51 children) and France (12 children) indicated that InO was well tolerated and effective in children with R/R BCP-ALL.15-17 The current phase 1 investigation (part of an approved Pediatric Investigational Plan18 ) prospectively evaluated the safety and tolerability, efficacy, and pharmacokinetics (PK)/pharmacodynamics (PD) of InO monotherapy to identify a recommended phase 2 dose (RP2D) for pediatric patients with CD22+ R/R ALL.
Methods
Innovative Therapies for Children With Cancer in Europe-059 (ITCC-059) (Dutch Trial Registry: NTR5736) is a phase 1/2 multicenter, single-arm, open-label study of InO in childhood CD22+ R/R BCP-ALL. Here, we present results from the phase 1 single-agent dose-finding investigation.
The study was conducted in accordance with the International Ethical Guidelines for Biomedical Research Involving Human Subjects, International Conference on Harmonization Guidelines for Good Clinical Practice, the Declaration of Helsinki, and applicable local regulatory requirements and laws. The study protocol was approved by the sponsor (Erasmus Medical Center Sophia Children’s Hospital, Rotterdam, The Netherlands), and by the institutional review boards and ethics committees (IRBs/ECs) in all participating centers. Parents and/or patients provided written informed consent, and patients were enrolled between January 2017 and April 2019 at 11 sites of the ITCC consortium.19
Patients
Eligible patients were age ≥1 year to <18 years at enrollment, had a diagnosis of CD22+ R/R BCP-ALL, an M2 or M3 bone marrow status, and refractory disease or second relapse or greater, or any relapse after HSCT. Exclusion criteria included any history of previous or ongoing veno-occlusive disease or SOS (modified Seattle criteria).20 For further entry criteria see supplemental Table 1 (available on the Blood Web site).
Study design
The primary objective was to determine the maximum tolerated dose (MTD) or RP2D of intravenous (IV) InO monotherapy; secondary objectives included safety, response, and PK/PD. A modified Rolling-6 escalation design21 was used, with dose de-escalation based on dose-limiting toxicities (DLTs) during course 1. In the first cohort, 2 patients at dose level 2 (DL2) experienced a DLT per protocol definition. Upon steering committee review, 1 of these toxicities (ie, aminotransferase elevation) was not considered to have significantly impaired patient safety (see supplemental Table 2 for details), and an IRB/EC-approved amendment allowed repeating DL1 and DL2 for DLT assessment in a second cohort of patients with adapted guidelines for aminotransferase monitoring.
A maximum of 6 courses was permitted (2 or 3 courses only before HSCT). A course of therapy was defined as 3 doses of InO (IV infusion for 60 minutes) once per week on days 1, 8, and 15. Course 1 was planned to last 22 days, and all subsequent courses were planned to last 28 days, with delays allowed up to 42 days. To avoid infusion-related adverse events (AEs), pretreatment with methylprednisolone 1 mg/kg IV (maximum 50 mg) before each infusion of InO was recommended.
InO dose levels were assigned at study entry (Table 1). In course 1, DL1 was 1.4 mg/m2 (80% of the adult dose) administered in a fractionated fashion, with the day 1 dose (0.6 mg/m2) higher than the following 2 doses (0.4 mg/m2), given the high tumor burden at relapse. In addition, preclinical studies suggest that fractionated schedules of lower InO doses compared with a single-course high dose may improve anti-ALL activity and reduce toxicities.16,22
InO dose escalation and de-escalation schedule
| Dose level | IV InO dose (mg/m2) | |||||||
|---|---|---|---|---|---|---|---|---|
| Course 1 | Courses 2-6* | |||||||
| Day 1 | Day 8 | Day 15 | Total doses per course | Day 1 | Day 8† | Day 15† | Total doses per course | |
| –2 | 0.4 | 0.2 | 0.2 | 0.8 | 0.2 | 0.2 | 0.2 | 0.6 |
| –1 | 0.5 | 0.3 | 0.3 | 1.1 | 0.3 | 0.3 | 0.3 | 0.9 |
| +1 (start) | 0.6 | 0.4 | 0.4 | 1.4 | 0.4 | 0.4 | 0.4 | 1.2 |
| +2 | 0.8 | 0.5 | 0.5 | 1.8 | 0.5 | 0.5 | 0.5 | 1.5 |
| +3 | 1.0 | 0.6 | 0.6 | 2.2 | 0.6 | 0.6 | 0.6 | 1.8 |
| Dose level | IV InO dose (mg/m2) | |||||||
|---|---|---|---|---|---|---|---|---|
| Course 1 | Courses 2-6* | |||||||
| Day 1 | Day 8 | Day 15 | Total doses per course | Day 1 | Day 8† | Day 15† | Total doses per course | |
| –2 | 0.4 | 0.2 | 0.2 | 0.8 | 0.2 | 0.2 | 0.2 | 0.6 |
| –1 | 0.5 | 0.3 | 0.3 | 1.1 | 0.3 | 0.3 | 0.3 | 0.9 |
| +1 (start) | 0.6 | 0.4 | 0.4 | 1.4 | 0.4 | 0.4 | 0.4 | 1.2 |
| +2 | 0.8 | 0.5 | 0.5 | 1.8 | 0.5 | 0.5 | 0.5 | 1.5 |
| +3 | 1.0 | 0.6 | 0.6 | 2.2 | 0.6 | 0.6 | 0.6 | 1.8 |
After course 1, in patients who have achieved a CR, the day 1 dose is decreased slightly because of no loading dose requirement. In patients who have not yet achieved a CR after course 1, a loading dose similar to that in course 1 was given in course 2, but not in subsequent courses.
Visit window of ±1 day.
End points
The primary end point was the incidence of DLTs during the first course of therapy, defined as any of the following toxicities related to InO: any grade 5 toxicity; absolute neutrophil count <500/μL and/or a platelet count <50 000/μL lasting beyond day 42 in the absence of persisting leukemia, or grade 3 to 4 nonhematologic toxicities persisting for >48 hours (>7 days for liver test abnormalities).
Secondary end points were safety and tolerability, including AEs, death attributable to InO, cumulative incidence of nonrelapse mortality, and hepatic SOS during or after InO. Secondary efficacy end points (detailed in supplemental text) were measures of antileukemic activity, including overall response rate (ORR), MRD status,23,24 duration of response (DOR), and number of patients undergoing post-InO treatment, including consolidation with HSCT or chimeric antigen receptor (CAR) T-cell therapy. Other end points were serum PK of unconjugated calicheamicin and InO.
Statistical analysis
The dose escalation analysis set (evaluable for establishing MTD/RP2D) included all enrolled patients who received ≥1 dose of InO and experienced a DLT during course 1 or who received ≥2 doses of InO without DLTs during course 1. MTD/RP2D was the highest dose level tested at which ≤1 of 6 patients experienced DLTs during course 1, with ≥2 patients experiencing DLTs at the next higher dose. If the MTD was not reached at the highest dose level (DL3), there would be no further dose escalations. Instead, the highest tested dose would be taken forward as the RP2D. Event-free survival and overall survival (OS) were estimated by using the Kaplan-Meier method.
The full analysis set included all enrolled patients who received ≥1 doses of study drug and was used for the efficacy and safety analyses. The PK analysis set included all patients in the full analysis set who provide ≥1 blood samples for PK.
Results
Data cutoff date was March 4, 2020, with a median follow-up of 19 months (range, 2-21 months).
Patients
Overall, 27 patients consented and were screened for inclusion. Twenty-five patients were enrolled (2 screening failures, both with inadequate liver function), and 23 were included in the dose escalation analysis set (supplemental Figure 2). Two patients in the first cohort who received only 1 dose of InO were not evaluable for DLTs as defined in the protocol. Treatment discontinuation in these 2 patients was not related to DLTs but to graft-versus-host disease reactivation in one patient and to sepsis shortly after the first dose of InO in another patient.
Patient baseline characteristics are summarized in Table 2. A total of 130 doses (median, 6 doses per patient; range, 1-12 doses per patient) and 42 courses (median, 2 courses per patient; range, 1-4 courses per patient) were given. Four courses were incomplete (1-2 doses only).
Patient characteristics
| Characteristic | No. (%) (N = 25) |
|---|---|
| Median age, y (range) | 11 (1.7-16.9) |
| Age category, y | |
| >1 to ≤2 | 1 (4) |
| >2 to ≤6 | 4 (16) |
| >6 | 20 (80) |
| Sex | |
| Male | 17 (68) |
| Female | 8 (32) |
| Bone marrow status at screening | |
| M3 | 22 (88) |
| M2 | 3 (12) |
| Median white blood cell count at screening × 109/L (range) | 3.5 (0.19-8.59) |
| Disease status at enrollment | |
| First relapse after HSCT | 7 (28) |
| Second or greater relapse | 15 (60) |
| Refractory | 3 (12) |
| Median No. of previous treatments (range) | 2 (2-7) |
| Specific elements of previous treatment | |
| HSCT | 14 (56) |
| Blinatumomab | 6 (24) |
| CAR T-cell therapy | 1 (4) |
| CD22 expression at screening | |
| Median MFI for CD22+ ALL cells (range) | 2768 (505-8370) |
| % CD22+ blasts (range) | 98 (53-100) |
| Cytogenetic subtype* | |
| Hypodiploid | 4 (16) |
| Hyperdiploid | 13 (52) |
| t[1;19](q23;p13) | 2 (8) |
| t[4;11](q21;q23) | 1 (4) |
| Normal cytogenetics | 4 (16) |
| Not done | 1 (4) |
| Characteristic | No. (%) (N = 25) |
|---|---|
| Median age, y (range) | 11 (1.7-16.9) |
| Age category, y | |
| >1 to ≤2 | 1 (4) |
| >2 to ≤6 | 4 (16) |
| >6 | 20 (80) |
| Sex | |
| Male | 17 (68) |
| Female | 8 (32) |
| Bone marrow status at screening | |
| M3 | 22 (88) |
| M2 | 3 (12) |
| Median white blood cell count at screening × 109/L (range) | 3.5 (0.19-8.59) |
| Disease status at enrollment | |
| First relapse after HSCT | 7 (28) |
| Second or greater relapse | 15 (60) |
| Refractory | 3 (12) |
| Median No. of previous treatments (range) | 2 (2-7) |
| Specific elements of previous treatment | |
| HSCT | 14 (56) |
| Blinatumomab | 6 (24) |
| CAR T-cell therapy | 1 (4) |
| CD22 expression at screening | |
| Median MFI for CD22+ ALL cells (range) | 2768 (505-8370) |
| % CD22+ blasts (range) | 98 (53-100) |
| Cytogenetic subtype* | |
| Hypodiploid | 4 (16) |
| Hyperdiploid | 13 (52) |
| t[1;19](q23;p13) | 2 (8) |
| t[4;11](q21;q23) | 1 (4) |
| Normal cytogenetics | 4 (16) |
| Not done | 1 (4) |
All data are presented as No. (%) unless otherwise specified.
MFI, mean fluorescence intensity.
Patients can have both hypodiploidy and a translocation.
Toxicity
In course 1 (cohort 1), DLTs were experienced by 1 of 6 patients at DL1 (grade 4 alanine aminotransferase [ALT] elevation), so this dose level was cleared. At DL2, DLTs were experienced by 2 of 5 patients (grade 4 ALT elevation for >7 days; no hematologic recovery at day 42). On review, the Steering Committee decided that the hepatic toxicity (aminotransferase elevation) did not significantly impair patient safety, because the patient was in CR after course 1 and received all 3 doses without being clinically ill or hospitalized for AEs. After an IRB/EC-approved protocol amendment, a second cohort was added with more stringent aminotransferase monitoring and dose delay criteria in case of aminotransferase elevation at day 8 after the first dose of InO. With this protocol modification, there were no hepatic DLTs at DL1 or DL2 in the second cohort: 0 of 6 patients at DL1 and 1 of 6 patients at DL2 had a DLT (no hematologic recovery at day 42), confirming the safety of both dose levels (supplemental Table 2). Given the very high response rate reached at DL2 (85%, see "Efficacy") with MRD negativity, the Steering Committee decided that no further improvements were to be expected with a higher dose of InO, a conclusion supported by preliminary PK data. Therefore, considering the risk-benefit balance, as well as the consideration that DL2 was the approved dose in adults, doses were not escalated to DL3, and DL2 was declared the RP2D.
During the study, all treated patients had ≥1 AEs, most frequently fever (n = 16; 64%), decreased platelet count (n = 15; 60%), decreased neutrophil count (n = 14; 56%), vomiting (n = 12; 48%), and anemia (n = 11; 44%) (supplemental Table 3). Twenty-three patients had ≥1 grade 3 to 4 AEs (all onset during course 1; Figure 1). Four patients had grade 5 AEs (disease progression with fatal outcome, n = 2; lung infection 2 months after administration of a single dose of InO, n = 1; sepsis after HSCT, n = 1), all of which were considered unrelated or unlikely to be related to InO. Laboratory and hematologic abnormalities are summarized in supplemental Table 4.
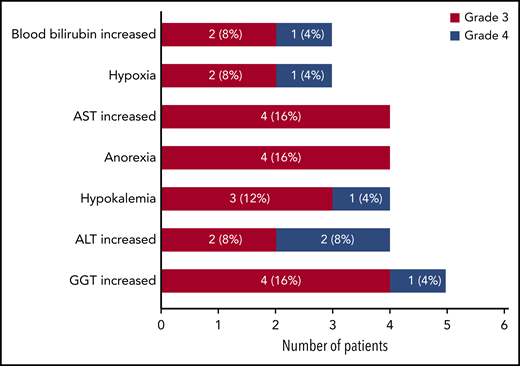
Most common nonhematologic AEs (grade 3 to 5; total >10%) reported as clinically significant by the investigators during the study (n = 25).The highest toxicity grade per patient was counted only once per patient. ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma-glutamyl transpeptidase.
Most common nonhematologic AEs (grade 3 to 5; total >10%) reported as clinically significant by the investigators during the study (n = 25).The highest toxicity grade per patient was counted only once per patient. ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma-glutamyl transpeptidase.
No cases of SOS were reported during InO treatment. However, during treatment after the study, two cases of SOS (grade 3 to 4, both ongoing at time of death as a result of infectious events) were recorded, both after subsequent multiagent chemotherapy (that included high-dose methotrexate and ifosfamide-cyclophosphamide) for R/R disease. One patient received 2 courses of InO at DL1, and another received 1 course at DL2, both without relevant signs of liver toxicity. Neither patient received HSCT before or after InO. No SOS occurred in the 7 patients who received a transplant after InO. These patients were treated with either 1 (n = 4) or 2 (n = 3) courses of InO. Only 1 of these patients received prophylaxis for SOS with defibrotide. Conditioning regimens included total body irradiation and etoposide (n = 3), and combined thiotepa and fludarabine with either treosulfan (n = 2), busulfan (n = 1), or melphalan (n = 1); 6 of 7 patients had not previously received a transplant.
Efficacy
ORR after course 1 was 80% (95% confidence interval [CI], 59% to 93%) for 20 of 25 patients: 75% (95% CI, 43% to 95%) at DL1 and 85% (95% CI, 55% to 98%) at DL2. Fifteen patients (60%) achieved CR, 1 patient achieved a CR with incomplete platelet recovery, and 4 patients achieved a CR with incomplete hematologic recovery (CRi). Characteristics of the 5 nonresponders (1 refractory, 2 with first relapse after HSCT, and 2 with a second or greater relapse) are summarized in supplemental Table 5.
Flow cytometry MRD data were available for 19 of 20 responding patients: 16 (84%) of 19 reached MRD-negative CR (bone marrow MRD <0.01%) as best response (6 of 9 patients at DL1 and 10 of 10 patients at DL2), of whom 14 were MRD negative after course 1 (6 of 9 at DL1 and 8 of 9 at DL2; 1 patient had missing data at DL2). Highly comparable MRD data were obtained using molecular analysis (supplemental Figure 3).
Median DOR after InO treatment was 8 months (range, 1-19 months); 6.9 months for patients treated at DL1 and 12.6 months for patients at DL2 (Figure 2; supplemental Table 7). Seven patients received consolidation treatment with HSCT (median, 51 days; range, 23 to 125 days after the last dose of InO), 4 without any bridging therapy, 3 after receiving additional chemotherapy (oral methotrexate or multiagent reinduction treatment) or blinatumomab before HSCT. Two patients received CAR T cells as consolidation treatment after achieving CR with InO (31 and 97 days after last dose of InO). Cells were harvested before treatment with InO in one patient and after treatment with InO in the other patient. Notably, 1 patient showed a prolonged continuous complete remission (CCR) (15 months) after 2 courses of InO without any additional treatment; 2 other patients were in CCR (both 19 months) without receiving HSCT or CAR T-cell therapy, but 1 patient did receive additional chemotherapy and the other patient received blinatumomab (Figure 2) after treatment with InO.
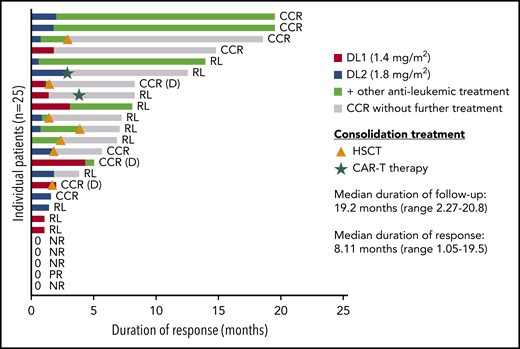
Duration of response to InO treatment. The figure shows the duration of response starting from the first achievement of CR. The green color of the bar highlights the start of other antileukemic treatment, and the gray portion shows the duration of response without additional treatment after the end of InO treatment. Consolidation treatment with HSCT or CAR T-cell therapy is also shown with symbols in the figure. CCR (D), death in CCR; NR, no response; PR, partial response; RL, relapse.
Duration of response to InO treatment. The figure shows the duration of response starting from the first achievement of CR. The green color of the bar highlights the start of other antileukemic treatment, and the gray portion shows the duration of response without additional treatment after the end of InO treatment. Consolidation treatment with HSCT or CAR T-cell therapy is also shown with symbols in the figure. CCR (D), death in CCR; NR, no response; PR, partial response; RL, relapse.
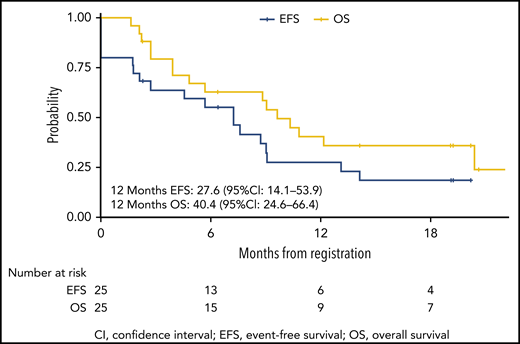
OS from the start of InO treatment was 63% (95% CI, 46% to 85%) at 6 months and 40% (95% CI, 25% to 66%) at 12 months (Figure 3). The median OS was reached at 7.2 months for patients treated at DL1, but it was not reached for DL2. EFS rate was 55% (95% CI, 39% to 79%) at 6 months and 28% (95% CI, 14% to 54%) at 12 months (Figure 3). Over the whole study, 15 patients either had no response (5 patients) or they relapsed. The cumulative incidence of nonresponse or relapse was 36% (95% CI, 18% to 55%) at 6 months and 59% (95% CI, 36% to 77%) at 12 months (supplemental Figure 1). Fifteen patients died during the study or during follow-up, including 3 nonrelapse deaths (2 were transplant-related and 1 was a result of multiorgan failure in aplasia after additional chemotherapy for MRD positivity). The cumulative incidence of nonrelapse mortality was 8% (95% CI, 1% to 24%) at 6 months and 13% (95% CI, 3% to 31%) at 12 months (supplemental Figure 1).
CD22 expression
All patients were CD22+ at enrollment, as confirmed at the central laboratory (Table 2). The difference in CD22 expression, determined both as mean fluorescence intensity and as percentage of blasts expressing CD22 antigen, was not statistically significant between responders and nonresponders (P > .05 for both [Wilcoxon test]). During follow-up, 13 patients had material available for reliable CD22 analysis at relapse: 2 of 13 were CD22−; in 1 additional patient, 80% of the residual blasts were CD22−. In 1 patient with KMT2A-rearranged, CD22 expression was still present at relapse, although 7% of the ALL blasts were CD22−. B-cell count was assessed at the end-of-treatment visit in 15 patients in CR: 3 patients had peripheral B-cell recovery at that time (median 21 days after last dose of InO); 1 additional patient reached B-cell recovery 3 months after the last dose of InO before receiving CAR T-cell therapy. Follow-up data are limited because of subsequent HSCT or relapse before recovery.
PK
The InO serum concentration vs time profile (compared with simulated adult PK data) is shown in Figure 4. InO serum peak and trough concentrations at various visits and treatment courses are summarized in supplemental Table 6. After a single dose (day 1, course 1), InO median maximum concentration at DL1 (n = 12) was 159 ng/mL and at DL2 (n = 13), it was 130 ng/mL compared with the simulated adult concentration of 204 ng/mL at a dose of 1.8 mg/m2 per course. After multiple doses (day 1, course 2), InO median maximum concentration at DL1 (n = 9) was 217 ng/mL, and at DL2 (n = 5), it was 246 ng/mL, which was comparable to the simulated adult concentration of 234 ng/mL at a dose of 1.8 mg/m2 per course (supplemental Table 2).
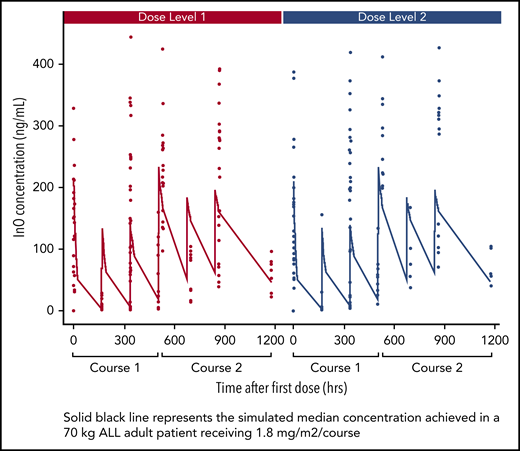
PK for InO serum concentration vs time profile in courses 1 and 2 by dose level.
PK for InO serum concentration vs time profile in courses 1 and 2 by dose level.
Discussion
In this dose-finding study, InO was well tolerated with notable efficacy in heavily pretreated children with R/R ALL. RP2D was established at 1.8 mg/m2 per course (0.8, 0.5, 0.5 mg/m2), as in adults.7 The ORR was 80%, with a 1-year OS probability of 40%. The response rate was higher for patients treated at DL2 (85%) compared with DL1 (75%), although this difference was not statistically significant because of the small sample size and for the MRD-negativity rate, which was 66% for DL1 and 100% for DL2 (n = 10). These better results are also reflected in the longer DOR reached by patients treated at DL2 (6.9 months at DL1 vs 12.6 months at DL2), as well as in the median survival (reached at 7.2 months for patients treated at DL1 and not reached at DL2) (supplemental Table 7).
Nine of 25 patients received consolidation therapy (HSCT or CAR T-cell therapy); 3 patients showed prolonged CCR without subsequent HSCT or CAR T-cell therapy (2 patients received additional therapy and 1 did not receive any treatment after InO) (Figure 2). Two cases of SOS were recorded after subsequent multiagent chemotherapy, as previously reported in non–transplant-related SOS25 ; no patient given HSCT developed SOS.
The disposition of InO after IV administration is influenced by interaction with CD22+ B cells and, as with other therapeutic proteins, in part to binding with the Fc receptor.26 The PK of InO in adults has been well characterized by a 2-compartmental model with linear and time-dependent clearance using pooled data from 765 adult patients.27 Steady-state InO levels were achieved after 3 to 4 cycles of treatment, with exposure increasing with treatment cycle (ie, clearance decreases over time). This is consistent with the PK of antibodies that target B-cell receptors showing target-mediated drug disposition as well as a half-life relative to the given dosage interval.27,28 This study is the first to include PK of InO in children with ALL. The peak and trough concentrations of InO after single and multiple doses were variably higher or lower in children when compared with the simulated data in a typical adult patient with ALL. Given the inconsistent direction of change (lower or higher) and considerable data variability, we concluded that InO plasma PK is comparable between pediatric and adult patients. Comparable plasma levels were also found in patients treated at DL1 and DL2. The comparable toxicity (supplemental Tables 3 and 4) and the results indicative of higher activity at DL2 (supplemental Table 7) led to the RP2D of 1.8 mg/m2 per course.
These preliminary data are from the first prospective analysis of InO in pediatric patients and are consistent with initial observations recorded in retrospective studies of InO in children, including a United States-European compassionate use program that reported a CR rate of 67%, and in that group of patients with CR, 71% achieved MRD negativity.15,16 Furthermore, there were no reports of SOS during treatment with InO in a retrospective, compassionate access study in 51 children; however, in contrast with this trial, SOS was reported in 11 (52%) of 21 patients after consolidation HSCT.15 These patients were heavily pretreated and received a median of 5 previous lines of therapy.15
In a retrospective post hoc analysis of this cohort, seven very-high-risk patients were identified (presenting with 1 or more of the following characteristics: low hypodiploidy, n = 2; MLL/AF4, n = 1; t(1;19) (q23;p13), n = 2; relapse <18 months after diagnosis, n = 6); of these, only 2 had no response to InO (1 with hypodiploidy and 1 with t[1;19] and very early relapse). No specific subgroups of patients with decreased efficacy were identified. Ongoing investigations in other cohorts of this study in R/R pediatric ALL include a phase 2 InO monotherapy cohort and a phase 1 dose-finding cohort of InO in combination with chemotherapy.
Preliminary results from the Children’s Oncology Group AALL1621 phase 2 study investigating InO monotherapy in a similar pediatric population at the adult dose, demonstrated a CR/CRi of 58%, with MRD <0.01% in 65% of responders and minimal hepatic toxicity during InO therapy; however, 4 of 13 patients who received a subsequent HCST (30.7%) developed SOS.29
InO data from a phase 3 randomized controlled study in 326 adults with R/R ALL showed that InO was superior to standard chemotherapy, with greater rates of CR/CRi (73.8% vs 30.9%; one-sided P < .0001).30 Similar response rates were observed in this pediatric study at the same dose established here as single-agent RP2D (1.8 mg/m2 per course). Subsequently, different dosages of InO have been tested in combination with chemotherapy regimens in adult ALL, and they show promising results.31,32
Two other immunotherapies are currently indicated for treatment of R/R BCP-ALL. Blinatumomab, a CD19-directed T-cell engager, and tisagenlecleucel, an anti-CD19 CAR T-cell therapy. The blinatumomab label for pediatric use33 was supported by a single-arm phase 1/2 study in pediatric patients with R/R ALL: 39% of patients achieved CR within 2 cycles of blinatumomab, 52% of whom achieved complete MRD response.34 The recent expanded access study showed even higher response rate (CR rate, 59%).35 Although not a direct comparison, these levels were lower than those observed with InO in this study. Recently, 2 courses of blinatumomab as post-reinduction therapy before HSCT showed superiority compared with 2 blocks of standard chemotherapy, leading to improved disease-free survival and OS.36 Tisagenlecleucel has been approved for treatment of patients age 25 years or younger with R/R ALL, based on findings from a phase 2 study that demonstrated an overall MRD-negative remission rate of 81% at 3 months.37,38 Because only 2 patients in this study received CAR T-cell therapy, no specific conclusions can be drawn on its use as consolidation therapy after InO. Although previous administration of InO should not lessen the efficacy of tisagenlecleucel, InO induces a posttreatment period of B-cell aplasia by targeting CD22 on both normal and malignant cells and, when there is MRD-negative CR plus B-cell aplasia, the expansion of CAR T cells may be impaired, potentially requiring a waiting time for B cells to reappear before CAR T-cell infusion.39,40 Other CD22-directed therapies are epratuzumab, an unconjugated CD22-directed antibody that showed limited activity,41,42 and CD22-directed CAR T-cell therapy.43
In summary, in this phase 1 study, InO was well tolerated and demonstrated antileukemic activity with high levels of MRD-negative response in children with R/R ALL, in line with observations in adults. The dose taken forward to the ongoing single-agent phase 2 cohort of this study was established as 1.8 mg/m2 (fractionated schedule) during course 1, as recommended in adults; the dose for subsequent courses remains at 1.5 mg/m2 per course up to a maximum of 6 courses unless HSCT is planned. Findings were consistent with adult PK analyses which indicated that the PK of InO is unaffected by age; thus, dose adjustment is not required in children. Dose-finding for InO in combination with 3-drug re-induction chemotherapy (dexamethasone, vincristine, PEG-asparaginase) in children with R/R ALL is ongoing.
Presented at the 61st Annual Meeting and Exposition of the American Society of Hematology, Orlando, FL, 7-10 December 2019.
Contact the corresponding author for original data. The study protocol is available to every investigator involved in the study. Data are regularly shared and discussed during investigator's meetings.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the Innovative Therapies for Children with Cancer (ITCC) Consortium and the International-BFM study group for providing the infrastructure and the collaborative environment to run early clinical trials in pediatric oncology, and Pfizer Inc for providing funding and a clinical research collaboration to implement the study. The authors also thank Berna Beverloo and the Department of Clinical Genetics, Erasmus Medical Center (Erasmus MC), for the central review of cytogenetic data. Medical writing support, under the direction of the authors, was provided by Juliet George, on behalf of CMC AFFINITY, McCann Health Medical Communications, in accordance with Good Publication Practice guidelines.
This study was sponsored by Erasmus MC Sophia Children’s Hospital, Department of Pediatrics, Rotterdam, The Netherlands, and was performed in a Clinical Research Collaboration and financially supported by Pfizer Inc. The KiKa Foundation provided infrastructural funding for the trial office at Erasmus MC and later at the Princess Máxima Center.
Authorship
Contribution: C.M.Z. conceived and designed the study; C.M.Z., A.C.J.A., E.B., V.H.J.v.d.V., L.V., F.L., K.N., C.C.-S., I.M.v.d.S., A.M., B. Brethon, I.Ø., L.S., C.D.-d.-H., B. Bielorai, C.R., and M.L.d.B. collected the data; C.M.Z., V.H.J.v.d.V., M.L.-Y., Y.C., E.B., A.T., and B.S. analyzed and interpreted the data; and all authors helped write the paper and review and approve the final version for submission.
Conflict-of-interest disclosure: F.L. was a member of the Board of Directors or Advisory Committee for Amgen, Bellicum, and Novartis; served as a consultant for Bluebird Bio, Bellicum, and Novartis; and received honoraria from Miltenyi and Amgen. C.D.-d.-H. was a member of the Advisory Committee or Speakers Bureau for Novartis and received honoraria from Amgen and travel funding from Jazz Pharmaceuticals and Novartis. C.R. served on the Advisory Boards for Amgen, Celgene, EUSA Pharma, Genentech, Novartis, and Roche and received honoraria from Bristol Myers Squibb, Pfizer, and Roche. V.H.J.v.d.V. received research funding from Jansen, BD Biosciences, and Amgen and honoraria from Amgen. I.M.v.d.S. served as a consultant for Jazz Pharmaceuticals and received research funding from Amgen, Servier, and Pfizer and travel funding from Jazz Pharmaceuticals. Y.C. and B.S. are employees of Pfizer Inc. B. Brethon served on the Advisory Board for Amgen and Astellas Pharma and received honoraria from Bristol Myers Squibb and Amgen. K.N. was a member of the Board of Directors or Advisory Committee for and served as a lecturer for Bayer and as a consultant for Y-mAbs Therapeutics. I.Ø. was a member of the Board of Directors or Advisory Committee for and served as a lecturer for Bayer. C.M.Z. served as a consultant for Servier, Sanofi, Daiichi Sankyo, Novartis, Janssen, Roche, Incyte, Pfizer, and Celgene; received research funding from Pfizer, Celgene, and Bristol Myers Squibb; and received travel funding from Jazz Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: Christian M. Zwaan, Princess Máxima Center for Pediatric Oncology, Heidelberglaan 25, 3584CS, Utrecht, The Netherlands; e-mail: c.m.zwaan@prinsesmaximacentrum.nl.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal