

TO THE EDITOR:
Somatic genetic rescue (SGR) is a rare spontaneous event in Mendelian diseases in which an in vivo genetic somatic mutation occurs in a given cell, conferring selective advantage and partially or totally annulling the effect of a pathogenic germline mutation.1 This phenomenon is observed in several inherited bone marrow failure syndromes, including Fanconi anemia,2 telomeropathies,3,4 and Shwachman-Diamond syndrome,5 and is usually associated with hematopoietic recovery. The molecular mechanisms leading to SGR are variable, from the reversion of the original pathogenic mutation (back mutation) or other site-specific mutations in the affected gene, to compensatory mutations in other genes and structural chromosome changes.1
GATA-binding protein-2 (GATA2) is a transcription factor that contains 2 zinc fingers and a nuclear localization signal that binds to W/GATA/R motifs.6 GATA2 is critical for the development of hematopoietic stem cells (HSCs) and for the establishment of definitive hematopoiesis.7 In animal models, GATA2 haploinsufficiency results in qualitative impairment in HSC function7 and, in humans, heterozygous germline mutations in GATA2 are etiologic in GATA2 deficiency, a pleomorphic disorder affecting hematopoiesis. It is characterized by monocytopenia, B- and NK-cell lymphopenia, bone marrow failure, myelodysplastic syndrome, and evolution to acute myeloid leukemia, as well as a myriad of extrahematopoietic clinical features, including disseminated nontuberculosis mycobacterial infections, warts, lymphedema, sensorineural hearing loss, and pulmonary alveolar proteinosis.6
To the best of our knowledge, SGR has not been reported in GATA2 deficiency. We describe for the first time the occurrence of SGR in the hematopoietic tissue in an asymptomatic individual carrying a heterozygous pathogenic germline mutation in GATA2 that generates a truncated protein without the 2 zinc fingers.
We started the investigation by screening the GATA2 gene for disease-associated variants in 2 siblings with GATA2 deficiency (detailed methods are in the supplemental Material, available on the Blood Web site). A 20-year-old man (Figure 1A; patient II-1) presented with a history of recurrent gram-negative infections and pain and stiffness of peripheral joints. Complete blood counts and flow cytometry analysis showed pancytopenia with monocytopenia, B and NK cell depletion. Serum IgG2 and IgG4 levels were low. Bone marrow biopsy showed hypocellularity (10%) without dysplasia. He was diagnosed with transfusion-independent moderate aplastic anemia and psoriatic arthritis. His pancytopenia worsened over the following 5 years, requiring blood transfusions, and a new biopsy showed 50% cellularity with trilineage dysplasia (Figure 2). He died at the age of 27 of a pulmonary infection. Postmortem Sanger sequencing revealed a heterozygous variant in GATA2 (NM_001145661.2:c.216C>A; NP_001139133.1:p.Y72*; Figure 1B), causing a premature stop codon and a truncated protein lacking the 2 DNA-binding zinc fingers and the nuclear localization signal.8 No variant in this GATA2 locus has been described in either the Genome Aggregation Database (gnomAD)9 or the Online Archive of Brazilian Mutations (ABraOM).10 The diagnosis of GATA2 deficiency was established.11 His brother (Figure 1A; patient II-2) also was heterozygous for the GATA2 p.Y72* variant in leukocytes and skin fibroblasts (Figure 1B). He first presented at the age of 25 years with a history of recurrent pulmonary infections, hypothyroidism, deep vein thrombosis (mesenteric and femoral), and deafness (Figure 2A). Complete blood counts and flow cytometry analysis showed reduced numbers of monocytes and B and NK cells (Figure 2B). The parents were screened and, whereas the mother was wild-type, the 61-year-old father (Figure 1A; subject I-1) harbored a heterozygous silent GATA2 p.Y72Y (NM_001145661.2:c.216C>T; rs1254938356) variant at the same locus (Figure 1B). He was asymptomatic, with no evidence of hematologic or extrahematopoietic manifestations of GATA2 deficiency. His blood counts and lymphocyte subsets were within the normal range (Figure 2B).
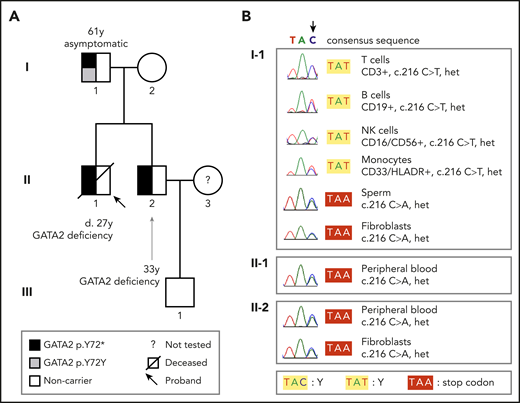
Pedigree and tissue-specific sequencing showing GATA2 mutations and disease phenotypes. (A) The heterozygous germline GATA2 p.Y72* mutation in affected brothers II-1 and II-2 segregated with GATA2 deficiency; the father (I-1) did not harbor this mutation in the blood by Sanger sequencing and was asymptomatic. (B) Sequencing chromatograms demonstrate that I-1 carried the GATA2 c.216 C>A p.Y72* mutation (as did II-1 and II-2) in sperm and fibroblasts, but not in blood subpopulations. Sorted T, B, and NK cells and monocytes from I-1 carried the silent somatic mutation in GATA2 c.216 C>T p.Y72Y, indicating somatic genetic rescue at the hematopoietic stem cell level. Het, heterozygous.
Pedigree and tissue-specific sequencing showing GATA2 mutations and disease phenotypes. (A) The heterozygous germline GATA2 p.Y72* mutation in affected brothers II-1 and II-2 segregated with GATA2 deficiency; the father (I-1) did not harbor this mutation in the blood by Sanger sequencing and was asymptomatic. (B) Sequencing chromatograms demonstrate that I-1 carried the GATA2 c.216 C>A p.Y72* mutation (as did II-1 and II-2) in sperm and fibroblasts, but not in blood subpopulations. Sorted T, B, and NK cells and monocytes from I-1 carried the silent somatic mutation in GATA2 c.216 C>T p.Y72Y, indicating somatic genetic rescue at the hematopoietic stem cell level. Het, heterozygous.
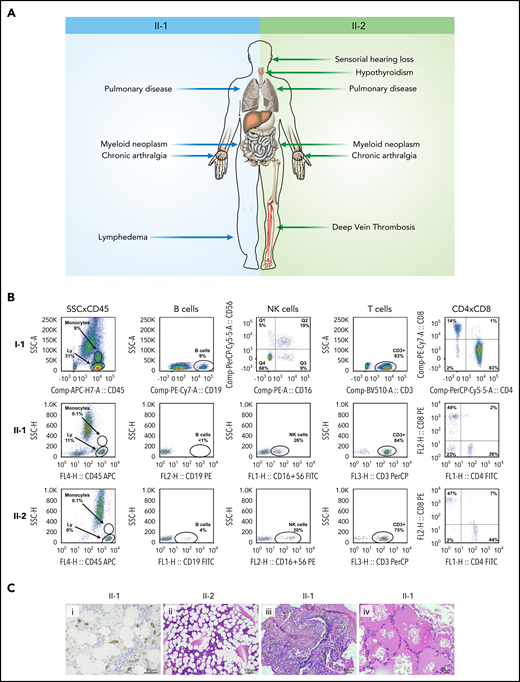
Clinical, laboratory, and pathologic features of patients with GATA2 deficiency. (A) Clinical findings in patients II-1 and II-2. (B) Immunophenotyping of the father (I-1), and brothers (II-1 and II-2) demonstrate normal peripheral blood counts in I-1 and monocytopenia and B-cell depletion in II-1 and II-2. (C) Bone marrow biopsy showing CD34+ clusters by immunohistochemistry in II-1 (i) and trilineage hypoplasia in II-2 (ii) and lung biopsy demonstrating chronic lymphoplasmacytic infiltrate (iii) and alveolar proteinosis (iv) in II-1. Hematoxylin and eosin stain. Ly, lymphocytes.
Clinical, laboratory, and pathologic features of patients with GATA2 deficiency. (A) Clinical findings in patients II-1 and II-2. (B) Immunophenotyping of the father (I-1), and brothers (II-1 and II-2) demonstrate normal peripheral blood counts in I-1 and monocytopenia and B-cell depletion in II-1 and II-2. (C) Bone marrow biopsy showing CD34+ clusters by immunohistochemistry in II-1 (i) and trilineage hypoplasia in II-2 (ii) and lung biopsy demonstrating chronic lymphoplasmacytic infiltrate (iii) and alveolar proteinosis (iv) in II-1. Hematoxylin and eosin stain. Ly, lymphocytes.
To elucidate the origin of the discordant variants in the same nucleotide, DNA was obtained from sperm and skin fibroblasts, which were found to carry the disease-associated GATA2 p.Y72* (c.216C>A) variant (Figure 1B), consistent with a site-specific mutation causing SGR in blood cells. Next-generation sequencing indicated mosaicism in the blood, as 93% of leukocytes harbored the somatic genetic variant (c.216C>T; p.Y72Y), whereas 7% carried the disease-associated c.216C>A p.Y72* variant (supplemental Figure 1A). To test whether SGR was skewed toward a specific subset, we sorted 4 cell types for sequencing: monocytes and T, B, and NK cells. The silent SGR variant was found in all lineages in heterozygosity (Figure 1B). As T cells have a long lifespan,12 the presence of SGR in T cells suggests that it occurred early in life. Also, that mosaicism involved B and NK cells indicates that SGR occurred at the HSC level, given that it has not been shown in pure B or NK cell immunodeficiency.1 Together, these results suggest that SGR occurs early in HSCs.
As SGR results from the selection and expansion of 1 of a few HSCs, proliferative stress may occur.13 To investigate, we measured the telomere length of peripheral blood leukocytes and found that the father’s telomere lengths were within the normal range for age (supplemental Figure 1B). However, patient II-2 showed rapid telomere erosion within 2 years of follow-up (supplemental Figure 1B). A single-nucleotide polymorphism array of the patients’ leukocytes showed uniparental disomy in chromosome 17q in patient II-2, but no chromosomal abnormality in the father (with SGR) or patient II-1 (supplemental Figure 1C). No myeloid neoplasm–associated somatic mutations or other germline variants were observed in the father or sons in next-generation sequencing of 154 genes associated with hematologic neoplasms and bone marrow failure syndromes (supplemental Material). These findings suggest that SGR derived from the expansion of a few HSCs promotes hematopoietic recovery.
Whereas the 2 sons presented severe extrahematologic complications (Figure 2), the father with SGR did not display any such phenotype. The stop codon generated by the pathogenic mutation deletes most of the GATA2 protein, including both zinc fingers, and is likely to result in clinical manifestations. That the father with SGR did not manifest clinical abnormalities in tissues retaining the pathogenic mutations suggests that early hematologic SGR and hematopoietic recovery prevented extrahematologic manifestations. It is known that bone marrow transplantation may alleviate pulmonary complications by restoring immunity and preventing recurrent infections.14 SGR may explain the paucity of symptoms in some patients carrying a disease-associated GATA2 variant and the variability in the age of onset.14,15 Also, SGR may cause false-negative results in presymptomatic or asymptomatic individuals tested in peripheral blood leukocytes. Geneticists should be aware of this possibility for genetic counseling and consider probing nonhematopoietic tissues. In the present case, hematologic SGR may have also influenced the absence of autoimmunity, lymphedema, and thrombosis. However, these observations need to be confirmed in further studies.
Finally, other molecular mechanisms may explain the heterogeneity in the clinical presentation. Using an allele-specific expression approach, Al Seraihi et al16 found that GATA2 monoallelic expression favoring the mutant allele is more frequent in symptomatic patients in comparison with biallelic expression (mutant and wild type) in asymptomatic individuals. They suggest that the expression ratio between alleles contributes to disease penetrance.
In aggregate, these results suggest that early recovery of hematopoiesis in GATA2 deficiency, either by transplantation or, potentially, by gene therapy, would be beneficial for hematopoiesis and would prevent other clinical complications.
The data reported in this article have been deposited in the National Center for Biotechnology Information (accession number PRJNA610566).
Original data are available by e-mail request to the corresponding author.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Fabiola Traina, Kamila Peronni Zueli, Alexandre T. Fabro, and Fernanda Borges da Silva for technical support, and Sandra Bresciani for her artwork assistance.
This study was supported by São Paulo Research Foundation (FAPESP) grants 13/08135-2 (L.F.B.C., A.L.P., D.V.C., B.A.S., and R.T.C.), 14/26379-9 (G.B.), and 17/09428-4 (F.S.D.).
Authorship
Contribution: L.F.B.C., G.B., D.V.C., and R.T.C. designed the study; L.F.B.C., G.B., A.L.P., and B.A.S. collected and prepared the samples collected from patients; L.F.B.C., G.B., A.L.P., F.S.D., F.C., and B.A.S. performed the experiments; L.F.B.C., G.B., A.L.P., F.S.D., F.C., and B.A.S. analyzed the data; L.F.B.C., F.S.D., B.A.S., and R.T.C. designed the figures; L.F.B.C., F.S.D., and R.T.C. wrote the manuscript; and R.T.C. supervised the conduct of the study.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Rodrigo T. Calado, Department of Medical Imaging, Hematology, and Clinical Oncology, FMRP-USP, Av. Bandeirantes, 3900 Laboratory Hematologia, bloco G, subsolo, HCRP, Ribeirão Preto, 14049-900, SP, Brazil; e-mail: rtcalado@usp.br.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal