

Key Points
A cell-based screening of 727 drugs in the NIH Clinical Collection library identified 9 drugs that may cause bleeding disorders.
Coagulopathy from drugs that inhibit the reduction of vitamin K epoxide, but not vitamin K, can be rescued by administering vitamin K.

Abstract
Drug-induced bleeding disorders contribute to substantial morbidity and mortality. Antithrombotic agents that cause unintended bleeding of obvious cause are relatively easy to control. However, the mechanisms of most drug-induced bleeding disorders are poorly understood, which makes intervention more difficult. As most bleeding disorders are associated with the dysfunction of coagulation factors, we adapted our recently established cell-based assay to identify drugs that affect the biosynthesis of active vitamin K–dependent (VKD) coagulation factors with possible adverse off-target results. The National Institutes of Health (NIH) Clinical Collection (NCC) library containing 727 drugs was screened, and 9 drugs were identified, including the most commonly prescribed anticoagulant warfarin. Bleeding complications associated with most of these drugs have been clinically reported, but the pathogenic mechanisms remain unclear. Further characterization of the 9 top-hit drugs on the inhibition of VKD carboxylation suggests that warfarin, lansoprazole, and nitazoxanide mainly target vitamin K epoxide reductase (VKOR), whereas idebenone, clofazimine, and AM404 mainly target vitamin K reductase (VKR) in vitamin K redox cycling. The other 3 drugs mainly affect vitamin K availability within the cells. The molecular mechanisms underlying the inactivation of VKOR and VKR by these drugs are clarified. Results from both cell-based and animal model studies suggest that the anticoagulation effect of drugs that target VKOR, but not VKR, can be rescued by the administration of vitamin K. These findings provide insights into the prevention and management of drug-induced bleeding disorders. The established cell-based, high-throughput screening approach provides a powerful tool for identifying new vitamin K antagonists that function as anticoagulants.
Introduction
Adverse drug reactions (ADRs) are unintended responses to medications that occur when doses are administered for diagnosis, prophylaxis, or treatment.1 ADRs represent a significant public health problem and are one of the leading causes of hospitalizations and deaths worldwide.2-5 In a meta-analysis, Lazarou et al6 estimated that ∼100 000 deaths that occur in the United States annually are caused by ADRs, which are the fourth leading cause of death following ischemic cardiopathy, cancer, and stroke. Estimated annual deaths resulting from ADRs in European countries are 197 000, with a total ADR cost to society of €79 billion.7,8 ADRs are most frequently associated with antithrombotic and antibacterial agents.2,9 Bleeding disorders are one of the most common causes of ADR-related hospitalizations.10-12
Drug-induced bleeding disorders contribute to substantial morbidity, mortality, and cost to society.13,14 Antithrombotic agents are the common drugs that cause unintended bleeding.15 Oral anticoagulants, including vitamin K antagonists (VKAs) and direct oral anticoagulants (DOACs), are used for the prevention and treatment of thromboembolic disorders.16 VKAs (such as warfarin) inhibit the biosynthesis of functional vitamin K–dependent (VKD) coagulation factors, whereas DOACs are direct inhibitors of factor Xa or thrombin. Because of their known mechanisms of action on anticoagulation, bleeding episodes associated with oral anticoagulants are relatively easy to predict and control.11,17 It has been shown that 96% of VKA-associated ADRs and 68% of DOAC-associated ADRs are preventable and avoidable.2,18,19 However, the pathogenic mechanisms for other drug-related bleeding disorders are less clear; thus, they are more harmful and difficult to control.
For example, nonsteroidal anti-inflammatory drugs are among the drugs most commonly prescribed worldwide to relieve pain and reduce inflammation. However, the treatment and prevention of nonsteroidal anti-inflammatory drug–associated coagulopathy remains challenging because of their unknown pathogenic mechanisms.20 In addition, the use of selective serotonin reuptake inhibitors, which are commonly prescribed antidepressants, has been shown to increase the tendency of gastrointestinal bleeding episodes21 and intracranial hemorrhages.12,22 Drugs that cause bleeding complications have been reported in a wide range of classes from antibiotics23-27 to drugs used in the treatment of sleeping problems, high blood pressure, and cancer chemotherapy.28,29 However, the molecular mechanisms underlying the coagulopathy caused by these drugs remain elusive. A better understanding of the molecular target and pathogenic mechanisms of drug-induced bleeding would provide important clues to the prevention and treatment of life-threatening bleeding disorders.
As most bleeding disorders are associated with the dysfunction of coagulation factors and increasing drug-induced bleeding complications have been reported to off-target the vitamin K cycle,25,30-32 the goal of this study was to functionally screen drugs that affect the biosynthesis of VKD coagulation factors. We first tested the feasibility of using our recently established cell-based assay33 as a high-throughput screening approach to identifying drugs that may target VKD carboxylation. We used the NIH Clinical Collection (NCC) library, which covers phase 1 to phase 3 clinical trial drugs, as an example for the screening test. We then used different cell-based assays to explore the possible mechanisms of anticoagulation on the top-hit drugs and to determine whether vitamin K plays a role in the reversal of this effect. Finally, we used a mouse model to test the bleeding risks of the selected top-hit drugs; 9 of the 727 drugs in the NCC library could function as VKAs, by inducing bleeding disorders. Both cell-based and animal model studies suggest that coagulopathy, which is caused by off-target reduction of vitamin K epoxide (KO) but not vitamin K, can be rescued with the administration of vitamin K.
Methods
Materials and cell lines
The NCC library was purchased from Evotec Inc (Princeton, NJ) as 2 sublibraries, 1 containing 446 drugs and the other, 281 drugs. All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO) or Cayman Chemical (Ann Arbor, MI). The monoclonal antibody against carboxylated glutamate (Gla) residues was from Sekisui Diagnostics, LLC (Stamford, CT). The GAPDH mouse monoclonal antibody was from Proteintech Group, Inc (Rosemont, IL). Human embryonic kidney 293 (HEK293) cells and human hepatoma G2 (HepG2) cells were from ATCC (Manassas, VA). HEK293 cells that stably express the reporter protein FIXgla-PC (protein C with its Gla domain replaced with that of factor IX) were created as previously described.34 FIXgla-PC/HEK293 cells with their endogenous vitamin K epoxide reductase (VKOR) and VKOR-like enzyme knocked out (double gene knockout, DGKO) were obtained by transcription activator‐like effector nucleases (TALENs)-mediated genome editing.35 HEK293 cells with the endogenous γ-glutamyl carboxylase (GGCX) knocked out were created by clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9–mediated genome editing.36
Cell-based VKD carboxylation activity assay
The cell-based VKD carboxylation activity assay was performed in FIXgla-PC/HEK293 cells with a convenient enzyme-linked immunosorbent assay (ELISA),34 which can be easily adapted to high-throughput screening. To differentiate which enzymatic activity of the vitamin K redox cycle is targeted by a given VKA, we used FIXgla-PC/HEK293 and DGKO cells, combined with KO or vitamin K as the substrate.
Cell-based VKOR activity assay
The cell-based VKOR activity assay was performed by incubating FIXgla-PC/HEK293 cells with KO.37 To evaluate the effect of a given drug on VKOR activity, we incubated FIXgla-PC/HEK293 cells with a fixed concentration or increasing concentrations of the drug in cell culture medium containing 5 µM KO for 24 hours. It should be noted that, to be an active cofactor for VKD carboxylation, KO must be reduced to vitamin K hydroquinone (KH2) by a 2-step process (Figure 1A). Drugs that affect any of the enzymatic activities for vitamin K redox cycling will affect the final readout of the assay. Thus, this assay is also used for the high-throughput screening of the NCC library.
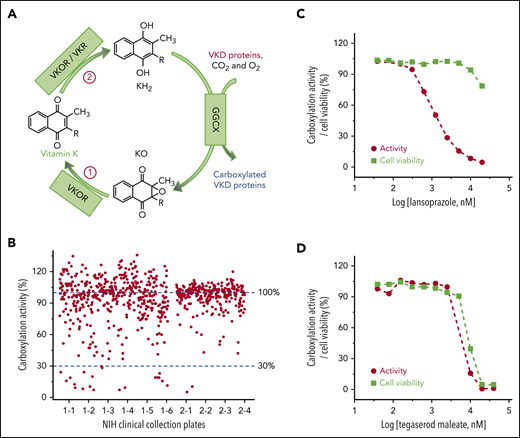
Cell-based screening of the NCC library for drugs that off-target the vitamin K cycle. (A) Vitamin K redox cycle. When VKD proteins are carboxylated by GGCX, by using vitamin K hydroquinone (KH2), carbon dioxide (CO2), and oxygen (O2) as cofactors, KH2 is oxidized to KO, which must be reduced to KH2 by a 2-step reduction. (B) Functional screenings of the 727 drugs in the NCC library. FIXgla-PC/HEK293 cells were incubated with the drugs identified in the NCC library (final concentration, 10 µM) in cell culture medium containing 5 µM KO. After a 48-hour incubation, reporter protein carboxylation was evaluated by ELISA. Carboxylation activity of DMSO-treated cells was normalized to 100%. The effect of lansoprazole (C) and tegaserod maleate (D) on VKD carboxylation and cell viability. FIXgla-PC/HEK293 cells were incubated with increasing concentrations of the test drug in cell culture medium containing 5 µM KO. Twenty-four hours later, cell culture medium was used for the cell-based activity assay and the drug-treated cells were used for the cell viability assay.
Cell-based screening of the NCC library for drugs that off-target the vitamin K cycle. (A) Vitamin K redox cycle. When VKD proteins are carboxylated by GGCX, by using vitamin K hydroquinone (KH2), carbon dioxide (CO2), and oxygen (O2) as cofactors, KH2 is oxidized to KO, which must be reduced to KH2 by a 2-step reduction. (B) Functional screenings of the 727 drugs in the NCC library. FIXgla-PC/HEK293 cells were incubated with the drugs identified in the NCC library (final concentration, 10 µM) in cell culture medium containing 5 µM KO. After a 48-hour incubation, reporter protein carboxylation was evaluated by ELISA. Carboxylation activity of DMSO-treated cells was normalized to 100%. The effect of lansoprazole (C) and tegaserod maleate (D) on VKD carboxylation and cell viability. FIXgla-PC/HEK293 cells were incubated with increasing concentrations of the test drug in cell culture medium containing 5 µM KO. Twenty-four hours later, cell culture medium was used for the cell-based activity assay and the drug-treated cells were used for the cell viability assay.
Cell-based vitamin K reductase activity assay
In general, vitamin K reductase (VKR) activity can be determined by incubating FIXgla-PC/HEK293 cells with 5 µM vitamin K. To eliminate the contribution of VKOR to vitamin K reduction,38 we used DGKO cells for the VKR activity assay.
Cell viability assay
The cytotoxicity of the test drugs was evaluated using a CCK-8 kit (Dojindo Molecular Technologies, Inc, Rockville, MD) according to the manufacturer’s instructions.
Cell-based KO and vitamin K reduction activity assay
Enzymatic activity of the 2-step vitamin K reduction was also evaluated in HEK293 cells by directly measuring different forms of vitamin K, with a conventional reversed-phase high-performance liquid chromatography (HPLC) assay.39
Cell-based KO reduction activity assay
GGCX-knockout HEK293 cells were incubated with 10 µM KO in the presence or absence of a 10-µM test drug for 5 hours. K vitamins were extracted from the treated cells and quantified by reversed-phase HPLC.39,40 We used GGCX-knockout HEK293 cells to prevent the conversion of the product (vitamin K) back to the substrate (KO) by the cell’s endogenous enzyme (Figure 1A).
Cell-based vitamin K reduction activity assay
DGKO cells were incubated with 10 µM vitamin K and followed by the same procedures as described above for the KO reduction activity assay. Because reduced vitamin K (KH2), an intermediate product in VKD carboxylation, is unstable and difficult to quantify accurately directly from cells,41 we coupled the vitamin K reduction and epoxidation reactions to measure the final stable product KO (Figure 1A). To ensure that the product (KO) is not reduced to the substrate (vitamin K) by the cells’ endogenous VKOR, we used DGKO cells in this assay.
Cell-based, high-throughput screening of the NCC library
The NCC library drugs were first diluted to 400 µM with dimethyl sulfoxide (DMSO), to make stock plates. Then, 5 µL of the diluted stock solution was transferred to 96-well plates and mixed with 195 µL of FIXgla-PC/HEK293 cells in growth medium containing 5 µM KO. The final screening concentration of drugs (10 µM) is higher than the physiological concentration of most drugs, to ensure that the preliminary screenings would not miss drugs with low inhibition efficiency. We used DMSO and warfarin as negative and positive controls, respectively, during the screening.
Anticoagulation test in an animal model
To eliminate any effect of sex on the coagulation test,42,43 we used only male mice in this study. Eight-week-old BALB/c male mice were acclimated for 1 week and were randomly collected into groups of 10. Two groups were treated with 1 drug, and 1 group was given vitamin K–supplemented (10 mg/L) drinking water. Mice were treated with either the drugs or the control DMSO for 7 consecutive days by intragastric administration, using dosages of 3 mg/kg per day warfarin, 400 mg/kg per day itraconazole, 100 mg/kg per day clofazimine, or 500 mg/kg per day nitazoxanide. The dosages are based on the inhibition potency of each drug for VKD carboxylation (Table 1) and on dosages from previous studies (supplemental Table 1, available on the Blood Web site). Twenty-four hours after the final treatment, the mice were euthanized, and blood samples were collected. The plasma sample was prepared by centrifugation at 3000g for 10 minutes at 4°C. Prothrombin times (PTs) were determined by Beijing Sino-UK Institute of Biological Technology (Beijing, China). All animal protocols were approved by the Animal Care and Use Committee of the Institute of Medicinal Plant Development at the Chinese Academy of Medical Sciences.
IC50and possible targets of the 9 top-hit drugs on vitamin K–dependent carboxylation
| Drugs | IC50 ± SD, nM | Potential target* | Increased INR or bleeding risks† | |
|---|---|---|---|---|
| KO reduction | K reduction | |||
| Warfarin | 8 ± 1 | >10 000 | KO reduction | Yes |
| Nitazoxanide | 124 ± 9 | >10 000 | KO reduction | Yes |
| Lansoprazole | 967 ± 49 | >10 000 | KO reduction | Yes |
| Itraconazole | 1 161 ± 157 | 258 ± 21 | Vitamin K availability | Yes |
| Clofazimine | 789 ± 91 | 2 964 ± 669 | Vitamin K reduction | Yes |
| Idebenone | 1 858 ± 408 | 2 196 ± 621 | Vitamin K reduction | — |
| AM404 | 1 976 ± 590 | 2 864 ± 621 | Vitamin K reduction | — |
| Nelfinavir | 8 305 ± 1 574 | 9 418 ± 1 234 | Vitamin K availability | Yes |
| Orlistat | >10 000 | >10 000 | Vitamin K availability | Yes |
| Drugs | IC50 ± SD, nM | Potential target* | Increased INR or bleeding risks† | |
|---|---|---|---|---|
| KO reduction | K reduction | |||
| Warfarin | 8 ± 1 | >10 000 | KO reduction | Yes |
| Nitazoxanide | 124 ± 9 | >10 000 | KO reduction | Yes |
| Lansoprazole | 967 ± 49 | >10 000 | KO reduction | Yes |
| Itraconazole | 1 161 ± 157 | 258 ± 21 | Vitamin K availability | Yes |
| Clofazimine | 789 ± 91 | 2 964 ± 669 | Vitamin K reduction | Yes |
| Idebenone | 1 858 ± 408 | 2 196 ± 621 | Vitamin K reduction | — |
| AM404 | 1 976 ± 590 | 2 864 ± 621 | Vitamin K reduction | — |
| Nelfinavir | 8 305 ± 1 574 | 9 418 ± 1 234 | Vitamin K availability | Yes |
| Orlistat | >10 000 | >10 000 | Vitamin K availability | Yes |
If a drug targets multiple steps in the vitamin K redox cycle, the most severe inhibition is listed.
References for increased INR and bleeding risks associated with each drug are listed in supplemental Table 2.
Results
Cell-based, high-throughput screening of the NCC library for VKAs
Vitamin K is an essential cofactor for the biosynthesis of VKD coagulation factors. Drugs that affect the redox cycling of vitamin K (Figure 1A) decrease the production of active coagulation factors, functioning as VKAs. To identify drugs in the NCC library that potentially function as VKAs, we incubated FIXgla-PC/HEK293 cells with 10 µM of the drug and 5 µM of KO to determine the carboxylation efficiency of the reporter protein. Most of the drugs in the NCC library do not affect reporter protein carboxylation (Figure 1B). However, 22 drugs decreased reporter protein carboxylation to less than 30% (supplemental Table 2). Warfarin, the most widely prescribed oral anticoagulant, was one of the top-hit drugs that abolished reporter protein carboxylation.
Decreased carboxylation of the reporter protein in the screening could also be related to the cytotoxicity of the test drugs. To exclude this possibility, we determined reporter protein carboxylation and cell viability using increasing concentrations of the 22 top-hit drugs. Lansoprazole significantly inhibited reporter protein carboxylation, but showed only minor cytotoxicity at higher concentrations (Figure 1C). This result suggests that a decrease in reporter protein carboxylation by lansoprazole is caused by an inhibition of VKD carboxylation rather than cytotoxicity. In contrast, the response curve of reporter protein carboxylation to tegaserod maleate closely follows the cell viability curve (Figure 1D), suggesting that the inhibition of reporter protein carboxylation by tegaserod maleate is mainly caused by its cytotoxicity or its effect on both carboxylation and cell viability, as described in "Discussion." Overall, of the 22 top-hit drugs (supplemental Figure 1), 9 may function as VKAs to inhibit VKD carboxylation (Table 1). The decreased production of the carboxylated reporter protein does not appear to be driven by a reduction in the synthesis of the reporter protein (supplemental Figure 2).
Identification of drugs targeting VKOR for KO reduction
The anticoagulation effect of VKAs could result from an inhibition of either the reduction of KO or vitamin K (Figure 1A). To explore the feasibility of using our cell-based assay to differentiate the reduction between KO and vitamin K, we performed warfarin titration of FIXgla-PC/HEK293 cells, using either KO or vitamin K as a substrate. When KO was used as the substrate, warfarin significantly inhibited VKD carboxylation (Figure 2A). However, when vitamin K was the substrate, warfarin showed only partial inhibition, which presumably resulted from the contribution of the warfarin-sensitive vitamin K reduction by VKOR.38,44 The contribution of vitamin K reduction by VKOR was further confirmed by using DGKO cells for the assay, which showed that warfarin no longer inhibited vitamin K reduction when VKOR was knocked out (Figure 2A). These results together suggest that, by using FIXgla-PC/HEK293 and DGKO reporter cells combined with KO or vitamin K as a substrate, we can identify drugs that target VKOR for KO reduction.
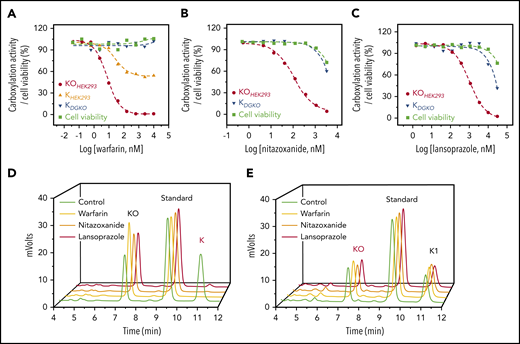
Characterization of drugs that inactivate VKOR for KO reduction. (A) Inhibition of VKD carboxylation by warfarin. FIXgla-PC/HEK293 cells were incubated with increasing concentrations of warfarin in cell culture medium containing 5 µM KO (KOHEK293) or vitamin K (KHEK293) for cell-based activity and cell viability assays, as described in the legend to Figure 1C. DGKO cells were used for a similar assay, with vitamin K serving as the substrate (KDGKO). Inhibition of VKD carboxylation by nitazoxanide (B) and lansoprazole (C). FIXgla-PC/HEK293 and DGKO cells were incubated with increasing concentrations of the test drug in cell culture medium containing 5 µM KO (KOHEK293) and vitamin K (KDGKO), respectively, for cell-based activity and cell viability assays. (D) Inhibition of KO reduction by warfarin, nitazoxanide, and lansoprazole in HEK293 cells. GGCX-knockout HEK293 cells were incubated with 10 µM KO in the presence or absence of 10 µM test drug for 5 hours. DMSO-treated cells were used as the control. KO reduction activity was evaluated by the production of vitamin K (red) in the chromatogram. (E) Inhibition of vitamin K reduction by warfarin, nitazoxanide, and lansoprazole in HEK293 cells. DGKO cells were incubated with 10 µM vitamin K in the presence or absence of 10 µM of the test drug for 5 hours. DMSO-treated cells were used as the control. Vitamin K reduction activity was evaluated by the production of KO (red) in the chromatogram.
Characterization of drugs that inactivate VKOR for KO reduction. (A) Inhibition of VKD carboxylation by warfarin. FIXgla-PC/HEK293 cells were incubated with increasing concentrations of warfarin in cell culture medium containing 5 µM KO (KOHEK293) or vitamin K (KHEK293) for cell-based activity and cell viability assays, as described in the legend to Figure 1C. DGKO cells were used for a similar assay, with vitamin K serving as the substrate (KDGKO). Inhibition of VKD carboxylation by nitazoxanide (B) and lansoprazole (C). FIXgla-PC/HEK293 and DGKO cells were incubated with increasing concentrations of the test drug in cell culture medium containing 5 µM KO (KOHEK293) and vitamin K (KDGKO), respectively, for cell-based activity and cell viability assays. (D) Inhibition of KO reduction by warfarin, nitazoxanide, and lansoprazole in HEK293 cells. GGCX-knockout HEK293 cells were incubated with 10 µM KO in the presence or absence of 10 µM test drug for 5 hours. DMSO-treated cells were used as the control. KO reduction activity was evaluated by the production of vitamin K (red) in the chromatogram. (E) Inhibition of vitamin K reduction by warfarin, nitazoxanide, and lansoprazole in HEK293 cells. DGKO cells were incubated with 10 µM vitamin K in the presence or absence of 10 µM of the test drug for 5 hours. DMSO-treated cells were used as the control. Vitamin K reduction activity was evaluated by the production of KO (red) in the chromatogram.
Like warfarin, nitazoxanide, and lansoprazole significantly inhibited VKD carboxylation in FIXgla-PC/HEK293 cells with KO as a substrate, but not in DGKO cells with vitamin K as a substrate, suggesting that nitazoxanide and lansoprazole mainly target VKOR (Figures 2B-C). To further confirm this, we determined KO and vitamin K reduction activities directly in HEK293 cells by using the conventional reversed-phase HPLC.39,40 Warfarin, nitazoxanide, and lansoprazole abolished KO reduction (Figure 2D), but had only a negligible effect on vitamin K reduction (Figure 2E), which is further support that these drugs mainly target VKOR for KO reduction.
Identification of drugs targeting VKR for vitamin K reduction
The characterization of idebenone (Figure 3A) and AM404 (supplemental Figure 3A) showed that the response of VKD carboxylation to these 2 drugs in FIXgla-PC/HEK293 cells overlapped with their responses in DGKO cells, suggesting that the knockout of VKOR has less effect on the inhibition of VKD carboxylation by these 2 drugs. Therefore, unlike warfarin, idebenone and AM404 presumably target VKR for vitamin K reduction. The inhibition of VKR by these 2 drugs is further supported by the results of our cell-based vitamin K reduction activity assay, which showed that idebenone and AM404 had only a moderate effect on KO reduction (Figure 3B; supplemental Figure 3B), but abolished the reduction of vitamin K (Figure 3C; supplemental Figure 3C).
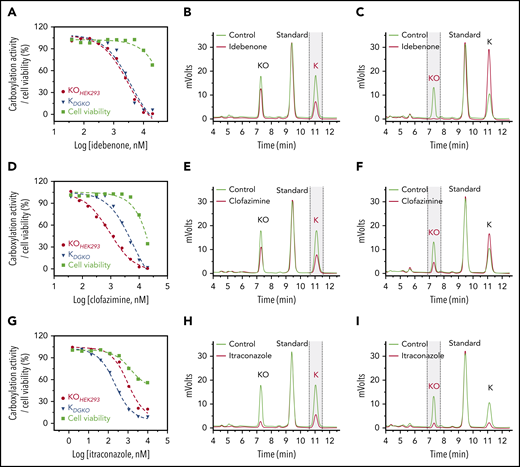
Characterization of drugs that inactivate VKR for vitamin K reduction or affect vitamin K availability within the cells. Inhibition of VKD carboxylation by idebenone (A), clofazimine (D), and itraconazole (G). FIXgla-PC/HEK293 and DGKO cells were incubated with increasing concentrations of the test drug in cell culture medium containing 5 µM KO (KOHEK293) and vitamin K (KDGKO), respectively, for the cell-based activity assay and cell viability assay (cell viability). Inhibition of KO reduction by idebenone (B), clofazimine (E), and itraconazole (H) in GGCX-knockout HEK293 cells. The KO reduction activity was evaluated by the production of vitamin K (marked with vertical dashed lines) in the chromatogram, as described in the legend to Figure 2D. Inhibition of vitamin K reduction by idebenone (C), clofazimine (F), and itraconazole (I) in DGKO cells. Vitamin K reduction activity was evaluated by the production of KO (marked with vertical dashed lines) in the chromatogram, as described in the legend to Figure 2E.
Characterization of drugs that inactivate VKR for vitamin K reduction or affect vitamin K availability within the cells. Inhibition of VKD carboxylation by idebenone (A), clofazimine (D), and itraconazole (G). FIXgla-PC/HEK293 and DGKO cells were incubated with increasing concentrations of the test drug in cell culture medium containing 5 µM KO (KOHEK293) and vitamin K (KDGKO), respectively, for the cell-based activity assay and cell viability assay (cell viability). Inhibition of KO reduction by idebenone (B), clofazimine (E), and itraconazole (H) in GGCX-knockout HEK293 cells. The KO reduction activity was evaluated by the production of vitamin K (marked with vertical dashed lines) in the chromatogram, as described in the legend to Figure 2D. Inhibition of vitamin K reduction by idebenone (C), clofazimine (F), and itraconazole (I) in DGKO cells. Vitamin K reduction activity was evaluated by the production of KO (marked with vertical dashed lines) in the chromatogram, as described in the legend to Figure 2E.
The effect of clofazimine on VKD carboxylation appears to be complex. FIXgla-PC/HEK293 cells were more sensitive to clofazimine inhibition than were DGKO cells, suggesting that clofazimine could have affected both KO and vitamin K reductions (Figure 3D). To clarify this point, we determined the effect of clofazimine on KO and vitamin K reductions by directly measuring K vitamins in the corresponding cells. Clofazimine affected both KO and vitamin K reductions (Figure 3E-F). It is worth noting that clofazimine and idebenone also had a minor effect on overall K vitamin levels in test cells (Figure 3B,E). This could result from their effect on either vitamin K entering the cells or metabolizing within the cells. We refer to this phenomenon as the effect of vitamin K availability within the cells. Nevertheless, these results suggest that idebenone, AM404, and clofazimine mainly target VKR for vitamin K reduction.
Characterization of other top-hit drugs in VKD carboxylation
Similar to clofazimine, VKD carboxylation in FIXgla-PC/HEK293 and DGKO cells had different sensitivities to itraconazole inhibition, suggesting that itraconazole affects both KO and vitamin K reductions (Figure 3G). Meanwhile, the results of directly measuring K vitamins in corresponding cells showed that itraconazole significantly affected vitamin K availability (Figure 3H-I). With such a low vitamin K concentration within the test cells, it was difficult to evaluate accurately the effect of itraconazole on KO or vitamin K reduction.
In contrast, orlistat and nelfinavir affected the overall VKD carboxylation, but appeared to have no effect on the enzymatic activity of vitamin K redox cycling (supplemental Figure 3D-I). The decreased VKD carboxylation by orlistat and nelfinavir were probably related to their effect on vitamin K availability, as it has been shown that orlistat reduces the absorption of fat-soluble vitamins, including vitamin K.45,46
The 9 top-hit drugs affected VKD carboxylation with different mechanisms. Bleeding complications associated with most of these top-hit drugs have been clinically reported (supplemental Table 2). The inhibition potency and the potential target of the 9 drugs on VKD carboxylation are presented in Table 1. Warfarin, nitazoxanide, and lansoprazole mainly targeted VKOR for KO reduction. Idebenone, clofazimine, and AM404 mainly inactivated VKR for vitamin K reduction. Itraconazole, and probably orlistat and nelfinavir, mainly affected vitamin K availability.
Inhibition of endogenous VKD protein carboxylation by the top-hit drugs
A caveat of our cell-based assay is that the exogenously overexpressed reporter protein may overload the reporter cells’ endogenous VKD carboxylation machinery, thus providing biased results. To test this possibility, we examined the carboxylation of the endogenous VKD proteins in HepG2 cells, a liver cell line that produces coagulation factors. We incubated the HepG2 cells with the individual drugs together with either KO or vitamin K. The carboxylation efficiency of the endogenous VKD proteins were detected by western blot analysis.47 Multiple carboxylated endogenous VKD protein bands of different sizes were detected when HepG2 cells were supplemented with KO or vitamin K (Figure 4; controls). Warfarin, nitazoxanide, and lansoprazole dramatically decreased the carboxylation of endogenous VKD proteins when HepG2 cells were incubated with KO (Fig. 4A), but had only a moderate effect on vitamin K–treated cells (Fig. 4B), consistent with VKOR being their primary target. Itraconazole, as we have shown, can have effects on vitamin K availability, and induced VKD carboxylation in both KO- and vitamin K–treated cells. The effects of idebenone, AM404, and clofazimine were somewhat unusual. For both KO- and vitamin K–treated cells, idebenone and AM404 had only minor effects on endogenous VKD protein carboxylation, whereas clofazimine significantly decreased VKD carboxylation. The various effects on VKD carboxylation between the VKR inhibitors may result from their different mechanisms of action, as described in "Discussion." In general, these results are consistent with results from the cell-based functional studies where the VKD reporter protein is overexpressed in the cells, suggesting that our cell-based assay is a useful tool for the functional study of VKD carboxylation.
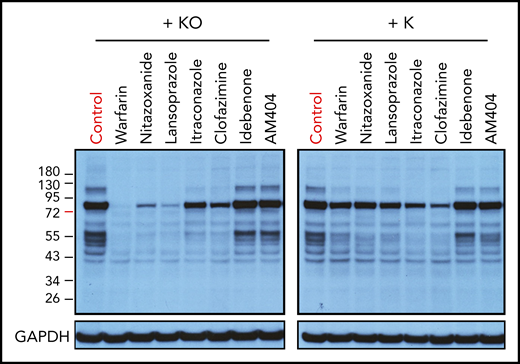
Effect of top-hit drugs on the carboxylation of endogenous VKD proteins. HepG2 cells were incubated with 3 µM of the drug in cell culture medium containing 5 µM KO (A) or vitamin K (B) for 24 hours. DMSO-treated cells were used as the control. The whole-cell lysate was used for western blot analysis with a monoclonal antibody that specifically recognizes carboxylated glutamate residues as the primary antibody. Cell lysates from an equal number of cells were loaded onto the gel, as demonstrated by the loading control of glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
Effect of top-hit drugs on the carboxylation of endogenous VKD proteins. HepG2 cells were incubated with 3 µM of the drug in cell culture medium containing 5 µM KO (A) or vitamin K (B) for 24 hours. DMSO-treated cells were used as the control. The whole-cell lysate was used for western blot analysis with a monoclonal antibody that specifically recognizes carboxylated glutamate residues as the primary antibody. Cell lysates from an equal number of cells were loaded onto the gel, as demonstrated by the loading control of glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
Reversal of the anticoagulant effect of the top-hit drugs by vitamin K
It is well-documented that the oral anticoagulant, warfarin, targets VKOR for KO reduction. This anticoagulation effect can be rescued by the administration of vitamin K that is directly reduced by VKR to support VKD carboxylation.48-52 However, if the anticoagulation effect of VKA results from the inhibition of VKR, this effect presumably cannot be rescued by administration of vitamin K (Figure 1A). To test this hypothesis, we incubated FIXgla-PC/HEK293 cells with each drug and with increasing concentrations of vitamin K. As expected, high concentrations of vitamin K recovered the reporter protein carboxylation to ∼100% in warfarin-, nitazoxanide-, and lansoprazole-treated cells (Figure 5A). However, vitamin K recovered only up to 60% carboxylation activity in idebenone-, clofazimine-, AM404-, and itraconazole-treated cells.
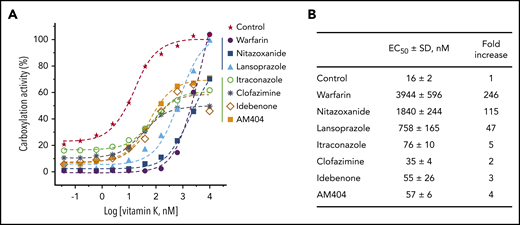
Reversal of the anticoagulation effect of the top-hit drugs by vitamin K. (A) FIXgla-PC/HEK293 cells were incubated with increasing concentrations of vitamin K in cell culture medium containing 3 µM of the test drug or DMSO (control) for 24 hours. The carboxylation efficiency of the reporter protein was determined by ELISA. (B) EC50 of vitamin K for reporter protein carboxylation in the presence of the test drugs or DMSO (control).
Reversal of the anticoagulation effect of the top-hit drugs by vitamin K. (A) FIXgla-PC/HEK293 cells were incubated with increasing concentrations of vitamin K in cell culture medium containing 3 µM of the test drug or DMSO (control) for 24 hours. The carboxylation efficiency of the reporter protein was determined by ELISA. (B) EC50 of vitamin K for reporter protein carboxylation in the presence of the test drugs or DMSO (control).
In addition, drugs that inactivate VKOR for KO reduction (warfarin, nitazoxanide, and lansoprazole) significantly increased the half-maximal effective concentrations (EC50) of vitamin K, whereas drugs that inactivate VKR for vitamin K reduction had only minor effects on EC50 (Figure 5A-B). We reasoned that the inactivation of VKOR blocks the reduction of KO to vitamin K. Therefore, enough vitamin K supplementation of the cell culture medium (increased EC50) will compensate for the inhibition of KO reduction. However, inactivation of VKR decreases the ability to produce KH2, the cofactor for VKD carboxylation (Figure 1A). Therefore, high vitamin K concentrations will not compensate for this defect.
Furthermore, a relatively high background signal was observed in the control cells and some of the cells treated with drugs that target vitamin K reduction, but not KO reduction (Figure 5A). This high background signal presumably results from VKOR recycling of the residual vitamin K in the cell culture medium, which has been confirmed by vitamin K titration in VKOR knockout cells (supplemental Figure 4).
Bleeding risks of the top-hit drugs in an animal model
To confirm the results from the above cell-based studies, we used a mouse model to examine the anticoagulation effect of the identified top-hit drugs. We selected itraconazole and clofazimine for the test, because bleeding complications have been clinically reported in the use of these 2 drugs.26,27,53,54 We also chose nitazoxanide for the animal test because, except for warfarin, it has the highest anticoagulation potency (Table 1). Similar to warfarin, all tested drugs have significantly increased PT (P < .01; Figure 6). Administration of vitamin K rescued the anticoagulation effect of warfarin and nitazoxanide, but not itraconazole and clofazimine. These results are consistent with results from the cell-based studies (Table 1; Figure 5A).
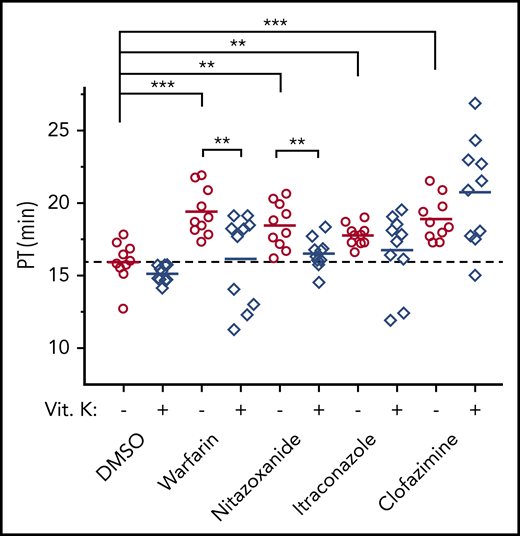
Anticoagulation effect of the selected top-hit drugs in a mouse model. BALB/c mice were treated with the test drugs for 7 consecutive days by intragastric administration, either with or without vitamin K–supplemented drinking water. Twenty-four hours after the final treatment, the PTs of the blood samples were determined. **P < .01; ***P < .001.
Anticoagulation effect of the selected top-hit drugs in a mouse model. BALB/c mice were treated with the test drugs for 7 consecutive days by intragastric administration, either with or without vitamin K–supplemented drinking water. Twenty-four hours after the final treatment, the PTs of the blood samples were determined. **P < .01; ***P < .001.
Discussion
The goal of this study was to establish a high-throughput approach for the screening of drugs that potentially cause bleeding disorders by off-target inhibition of vitamin K redox cycling. This requires a robust assay that is fast, reliable, easy to conduct, and sensitive enough to monitor the complicated VKD carboxylation reaction in its native environment. We reasoned that our recently established cell-based assays33 for the functional assessment of vitamin K cycle enzymes could be useful for achieving this goal. Therefore, we adapted our cell-based assay for use in the high-throughput screening of the 727 drugs in the NCC library that potentially cause bleeding risks.
Warfarin, the most widely prescribed oral anticoagulant, was the top candidate for bleeding risks in the screening. Characterization of warfarin for the inhibition of VKD carboxylation suggests that it targets VKOR for KO reduction (Figure 2), which is consistent with our knowledge that warfarin binds tightly near the active site of VKOR to inhibit its activity.55-59 Our results also suggest that, like warfarin, lansoprazole, and nitazoxanide mainly inactivate VKOR. Lansoprazole is a proton pump inhibitor that inhibits (H+/K+)-ATPase by forming disulfide bonds with the sulfhydryl groups of the parietal proton pump.60-62 This inhibition can be reversed by using reducing agents, such as dithiothreitol. Because VKOR uses 2 free cysteines (C132XXC135 redox center) for KO reduction,38,63 we assume that lansoprazole inactivates VKOR by blocking the sulfhydryl groups of its active site cysteines. We confirmed this hypothesis by the VKOR in vitro activity assay, showing that inactivation of VKOR by lansoprazole, but not warfarin, can be recovered by dithiothreitol (supplemental Figure 5). The inhibition of VKOR activity by nitazoxanide is probably associated with its function as an inhibitor of the protein disulfide isomerase,64 which has been proposed to be the physiological reductant of VKOR, serving as the electron donor for the active site regeneration of VKOR.65,66
However, it should be noted that drugs that affect the availability of electron donors for multiple-step reductions of vitamin K not only inhibit VKD carboxylation, but also could affect other important cellular functions, causing cytotoxicity. If this is the case, one would expect the inhibition curve of VKD carboxylation to closely follow the cell viability curve, as we observed in tegaserod maleate (Figure 1D) and for those in supplemental Figure 1. As the electron donors of the vitamin K cycle enzymes remain unknown, the mechanisms of inhibition for these drugs must be further clarified when the identity of these electron donors become available.
Characterization of idebenone and clofazimine on VKD carboxylation suggests that these 2 drugs mainly inhibit VKR for vitamin K reduction. It has been shown that clofazimine competes with menaquinone-4 (vitamin K2) for reduction.67,68 Idebenone, an analog of ubiquinone, can be efficiently reduced by NAD(P)H:quinone oxidoreductase 1,69 the enzyme that has been proposed to reduce vitamin K in VKD carboxylation.70,71 Therefore, both clofazimine and idebenone probably function as competitive inhibitors of vitamin K reduction to inhibit VKD carboxylation. In addition, reduced clofazimine is spontaneously reoxidized by O2 to release the reactive oxygen species.67,68 It has been shown that reactive oxygen species can neutralize reduced vitamin K (KH2),72,73 the cofactor of GGCX for VKD carboxylation. This additional effect of clofazimine on VKD carboxylation explains why clofazimine has a greater inhibitory potency than idebenone and AM404 for VKD carboxylation (Table 1; Figure 4). The molecular mechanisms of inhibition of VKD carboxylation by itraconazole, nelfinavir, and AM404 are not clear (Table 1). These drugs may not directly affect the enzymatic activity of the vitamin K redox cycle; rather, they may affect vitamin K availability within the cells, as some have been reported to reduce the absorption of fat-soluble vitamins, including vitamin K.45,46
Identifying drugs with off-target effects that inhibit vitamin K redox cycling will help in preventing and controlling bleeding disorders associated with ADRs. Functional screening of the NCC library identified 9 candidate drugs with bleeding risks that function as VKAs (Table 1). Most of these candidate drugs have been associated with bleeding diatheses or have an increased international normalized ratios (INRs) in patients (supplemental Table 2). However, the mechanisms underlying these bleeding events are unclear. Our results suggest that some of these drugs inhibit VKOR activity, and others inhibit VKR activity for the carboxylation of coagulation factors. Both cell-based and animal model studies show that if a drug inactivates VKOR, its anticoagulation effect can be rescued by vitamin K administration (Figures 5 and 6). However, if it inactivates VKR for vitamin K reduction, its anticoagulation effect will not be rescued by vitamin K. The differential inhibition of VKOR and VKR by these drugs is consistent with their distinct chemical structures (supplemental Figure 6), suggesting that these 2 enzymes possess different active sites for substrate and inhibitor binding. Although the mechanism of inhibition of VKOR by warfarin has been extensively studied by using biochemical approaches, molecular dynamics simulations, and molecular docking,55-57,74 no mechanistic study or computational modeling is available for the targeting of vitamin K cycle enzymes by other drugs. The results of this study have laid the foundation for exploring the mechanisms of action of other drugs impacting VKD carboxylation.
Clinical drug dosages are generally evaluated by the drug plasma concentrations. The half-maximal inhibition concentrations (IC50) of the identified top-hit drugs in this study are significantly smaller than (or close to) their known plasma concentrations, which supports the clinical observation of bleeding risks in patients who are using these drugs. For example, bleeding risks have been observed with the use of nitazoxanide at a daily dose of 500 mg, which has a drug plasma concentration (35 µM)75 of ∼300-fold higher than its IC50 (supplemental Table 1). On the other hand, the plasma concentration of itraconazole (1 µM)76,77 at a dosage that caused bleeding disorders is similar to its IC50. These dramatic variations between a drug’s plasma concentration and its IC50 may occur because coagulation factors are synthesized in the liver, which could have different drug concentrations than in the plasma. In addition, potential drug-drug interactions with other anticoagulants may cause bleeding risks at lower clinical dosages. For example, it has been reported that concomitant use of itraconazole78,79 and nelfinavir80 with warfarin elevates the INR and increases the bleeding risk of anticoagulation therapy. Nevertheless, our results suggest that when using these drugs in patients with anticoagulation therapy, clinicians should be aware of bleeding risks that result from off-target inhibition of the vitamin K cycle.
In summary, we have established a cell-based, high-throughput approach for screening drugs that have bleeding risks caused by off-target inhibition of vitamin K redox cycling. Functional screening of the 727 drugs in the NCC library identified 9 drugs that inhibit VKD carboxylation. Characterization of these drugs suggests that some drugs inhibit VKD carboxylation by off-targeting VKOR, whereas other drugs off-target VKR. Both cell-based and animal model studies suggest that the anticoagulation effects resulting from drugs that off-target VKOR, but not VKR, can be rescued by the administration of vitamin K. Results of this study provide insights into the molecular mechanisms of drug-induced bleeding as a result of off-target inhibition of the vitamin K cycle and provide clues on the prevention and management of related bleeding risks.
The authors will share the plasmid and the cell line used in this study in response to reasonable requests.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Lee Pedersen for helpful discussions and proofreading the manuscript.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grant HL131690 (J.K.T., D.W.S.); Chinese Academy of Medical Science (CAMS) Innovation Fund for Medical Sciences grant CIFMS 2016-12M-1-002 (K.H.); and Medical Epigenetics Research Center, Chinese Academy of Medical Sciences grant 2019PT310017 (K.H.).
Authorship
Contribution: X.C., B.I., D.W.S., K.H., and J.-K.T. conceived the experiments; X.C. performed the cell-based functional experiments; B.I. did the primary screening; D.-Y.J. and Z.H. created the reporter cell lines; C.L. and X.B. did the animal experiments; and X.C. and J.-K.T. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jian-Ke Tie, Department of Biology, The University of North Carolina at Chapel Hill, Chapel Hill, NC 27599; e-mail: jktie@email.unc.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal