

Key Points
Genetically determined γ′ fibrinogen is associated with lower risk of venous thromboembolism, cardioembolic stroke, and large artery stroke.

Abstract
Fibrinogen is a key component of the coagulation cascade, and variation in its circulating levels may contribute to thrombotic diseases, such as venous thromboembolism (VTE) and ischemic stroke. Gamma prime (γ′) fibrinogen is an isoform of fibrinogen that has anticoagulant properties. We applied 2-sample Mendelian randomization (MR) to estimate the causal effect of total circulating fibrinogen and its isoform, γ′ fibrinogen, on risk of VTE and ischemic stroke subtypes using summary statistics from genome-wide association studies. Genetic instruments for γ′ fibrinogen and total fibrinogen were selected, and the inverse-variance weighted MR approach was used to estimate causal effects in the main analysis, complemented by sensitivity analyses that are more robust to the inclusion of pleiotropic variants, including MR-Egger, weighted median MR, and weighted mode MR. The main inverse-variance weighted MR estimates based on a combination of 16 genetic instruments for γ′ fibrinogen and 75 genetic instruments for total fibrinogen indicated a protective effect of higher γ′ fibrinogen and higher total fibrinogen on VTE risk. There was also a protective effect of higher γ′ fibrinogen levels on cardioembolic and large artery stroke risk. Effect estimates were consistent across sensitivity analyses. Our results provide evidence to support effects of genetically determined γ′ fibrinogen on VTE and ischemic stroke risk. Further research is needed to explore mechanisms underlying these effects and their clinical applications.
Introduction
Fibrinogen is a key protein in the coagulation cascade that results in fibrin clots and in platelet aggregation.1 Fibrinogen is also part of the acute phase response, serving as an inflammatory mediator.2 The Aα, Bβ, and γ chains that comprise fibrinogen are encoded by the FGA, FGB, and FGG genes, respectively, which are located together in a single genetic region. Alternative splicing of FGG produces an isoform of the γ chain known as γ′, resulting in the production of γ′ fibrinogen, which makes up about 8% to 15% of total plasma fibrinogen.3,4 The last 4 amino acids encoded by exon 10 are replaced by the 20 amino acids encoded by intron 9, leading to the formation of γ′.5 The extension of γ′ fibrinogen contains a high affinity binding site for thrombin, allowing it to sequester thrombin, reducing thrombin’s ability to convert fibrinogen to fibrin.6 In addition, γ′ fibrinogen contains an extra binding site for the factor XIII B subunit7 and lacks the platelet integrin αIIbβ3 binding site found on the γ chain, which results in reduced platelet-fibrin(ogen) interactions.7
The FGG H2 haplotype, associated with lower levels of γ′ fibrinogen but not total fibrinogen, is associated with increased risk of venous thromboembolism (VTE).8 The association of variants in the FGG H2 haplotype with VTE has been recapitulated by large genome-wide association studies (GWASs), which have shown that it has a significant genetic association with VTE.9 More recently, a large GWAS has demonstrated that the same γ′ fibrinogen lowering variants are also associated with a higher risk of ischemic stroke.10
Results from the genetic studies on VTE and ischemic stroke could be explained by an overall anticoagulant effect of γ′ fibrinogen. Indeed, γ′ fibrinogen has several confirmed and suspected anticoagulant properties.6,7 However, the inverse association of γ′ fibrinogen with VTE and ischemic stroke has not been consistently reported in observational studies using case-control and prospective cohort approaches.11-15 In contrast, total fibrinogen levels are associated with an increased risk of ischemic stroke.16,17 A similar positive association with total fibrinogen levels can also be seen in some studies of VTE18,19 but not others.20 These contrasting associations may be because of the unique properties of the γ′ chain compared with the γ chain.
These observational studies may have been influenced by confounding and reverse causation. Mendelian randomization (MR) is an approach that uses genetic variants as instruments. Given that genetic variants are inherited in random and are constant over life, their associations will not be affected by confounding and reverse causation. To explore the causal effects of γ′ and total fibrinogen levels on VTE and ischemic stroke subtypes, we performed 2-sample MR, leveraging the results of large-scale GWASs. We also used MR to examine the effect of γ′ and total fibrinogen levels on D-dimer levels.
Methods
Genetic association estimates
The MR analyses used summary statistics from several GWASs, which are described in Table 1. All of the GWASs were restricted to European-ancestry participants, and all but one were based on genotypes imputed using the 1000 Genomes Project phase 1 version 3 reference panel. Analysis of the Atherosclerosis Risk in Communities (ARIC) study was approved by the University of Minnesota Institutional Review Board. All other datasets were summary statistics from published genome-wide association studies. Research was conducted in accordance with the Declaration of Helsinki.
Summary of genome-wide association studies used in the summary statistics–based 2-sample mendelian randomization analyses
| Phenotype | Sample size | Consortium/study |
|---|---|---|
| Exposures | ||
| Fibrinogen levels | 120 246 | CHARGE Hemostasis21 |
| γ′ fibrinogen levels | 9225 | ARIC study |
| Main outcomes | ||
| Venous thromboembolism | 7507 cases/52 632 controls | INVENT consortium9 |
| Any ischemic stroke | 34 217 cases/406 111 controls | MEGASTROKE10 |
| Cardioembolic stroke | 7193 cases/322 150 controls | MEGASTROKE10 |
| Large artery stroke | 4373 cases/204 991 controls | MEGASTROKE10 |
| Small vessel disease | 5386 cases/254 558 controls | MEGASTROKE10 |
| Additional phenotypes | ||
| C-reactive protein levels | 148 164 | CHARGE Inflammation22 |
| D-dimer levels | 21 052 | CHARGE Hemostasis23 |
| Phenotype | Sample size | Consortium/study |
|---|---|---|
| Exposures | ||
| Fibrinogen levels | 120 246 | CHARGE Hemostasis21 |
| γ′ fibrinogen levels | 9225 | ARIC study |
| Main outcomes | ||
| Venous thromboembolism | 7507 cases/52 632 controls | INVENT consortium9 |
| Any ischemic stroke | 34 217 cases/406 111 controls | MEGASTROKE10 |
| Cardioembolic stroke | 7193 cases/322 150 controls | MEGASTROKE10 |
| Large artery stroke | 4373 cases/204 991 controls | MEGASTROKE10 |
| Small vessel disease | 5386 cases/254 558 controls | MEGASTROKE10 |
| Additional phenotypes | ||
| C-reactive protein levels | 148 164 | CHARGE Inflammation22 |
| D-dimer levels | 21 052 | CHARGE Hemostasis23 |
We performed a GWAS of circulating γ′ fibrinogen concentration in European-ancestry participants from the ARIC study.24 The γ′ fibrinogen exposure (mg/dL) was measured in fasting citrated plasma collected at ARIC visit 3 using the enzyme-linked immunosorbent assay, as described previously.13 Levels of γ′ fibrinogen were natural log-transformed before analyses. Outliers with log γ' fibrinogen levels ± 3 standard deviations from the mean were excluded, resulting in a final sample size of 9225 participants. Genotypes were assessed using the Affymetrix (Santa Clara, CA) Genome-wide Human SNP Array 6.0 assay, and additional genotypes were imputed using IMPUTE225,26 based on the 1000 Genomes Project phase 1 version 3 reference panel.27 Genome-wide analyses were then performed using FAST version 1.8,28 adjusted for age, sex, and ancestry-informative principal components 1 and 2. A total of 9 335 343 variants with minor allele frequency >1% and imputation quality r2 > 0.3 were considered in the analysis.
To assess the effect of genetic variants on total fibrinogen levels, we used GWAS summary data from a published study of total circulating fibrinogen levels from the Cohorts for Heart and Aging Research in Genetic Epidemiology (CHARGE) Hemostasis Working Group.21 This study identified 41 genome-wide significant loci that explained 3% of variance in circulating plasma fibrinogen.21 To assess the effect of genetic variants on VTE, we used summary statistics from the largest published GWAS on VTE, performed by the International Network against VENous Thrombosis (INVENT) consortium.9 To assess the effect of genetic variants on ischemic stroke and its subtypes (cardioembolic stroke, large artery stroke, and small vessel disease) a GWAS by the MEGASTROKE consortium was used (Table 1).10 Cardioembolic stroke occurs when a clot in an artery of the heart forms and travels to obstruct a vessel in the brain.29 Large artery stroke usually results from a clot that forms in an atherosclerotic artery: when an atherosclerotic plaque ruptures, this triggers the formation of a blood clot that then travels to the brain and obstructs one of the large arteries.30 Small vessel disease is characterized by occlusion of the small vessels in the brain and reduced cerebral blood flow.31
Besides the exposures (γ′ fibrinogen and total fibrinogen levels) and clinical outcomes (VTE and ischemic stroke subtypes), we also used summary statistics from GWASs of 2 additional phenotypes. A GWAS of circulating D-dimer levels from the CHARGE Hemostasis Working Group23 was used so we could consider fibrin clot breakdown and its relationship with elevated circulating fibrinogen. The GWAS of D-dimer levels was based on genotypes imputed using the HapMap reference panel. We additionally used a GWAS from the CHARGE Inflammation Working Group of circulating C-reactive protein (CRP) level.22 Given the role of fibrinogen in inflammation,2 adjusting for the sensitive inflammatory marker CRP in the analyses allowed us to consider the role of inflammation in the associations of genetically determined fibrinogen with the clinical outcomes.
Genetic instruments
We separately applied the following steps to the summary statistics for γ′ fibrinogen and total fibrinogen levels to obtain sets of independent genetic instruments for each of these exposures: (1) we chose genetic instruments that were genome-wide significant (P < 5 × 10−8), (2) among the genome-wide significant variants, we chose those that were also present in the GWASs of the clinical outcomes, and (3) we used linkage disequilibrium (LD) clumping implemented in the TwoSampleMR R package to prune these variants according to their pairwise LD by retaining the variant with the most significant P value from each pair of variants with an LD of r2 > .1.32
Because the GWAS of D-dimer levels was based on genotypes imputed using the HapMap reference panel instead of the 1000 Genomes reference panel, not all selected instruments were present in the D-dimer summary statistics. We therefore repeated the steps above, restricting variants in step 2 to variants present in the summary statistics for D-dimer.
We calculated the proportion of variance in γ′ fibrinogen and total fibrinogen explained by each of the genetic instruments using the following formula: R2 = [β(x)2]/var(y) × 2 × EAF × (1 − EAF), where β(x) is the β coefficient for the association of the genetic instrument with the exposure, var(y) is the variance of the exposure, and EAF is the effect allele frequency of the genetic instrument. The variance of γ′ fibrinogen levels was calculated in the ARIC study, whereas for total fibrinogen, we calculated sample-size-weighted mean variance based on the variance in each of the studies that contributed to the total fibrinogen GWAS. The F statistic for each genetic instrument was then calculated using F = R2 × (N – 2)/(1 – R2).
Mendelian randomization: primary analysis
The TwoSampleMR R package was used to calculate estimates from genetic instruments of the causal effect of the exposure (γ′ fibrinogen or total fibrinogen levels) on each of the 5 clinical outcomes (VTE, any ischemic stroke, cardioembolic stroke, large artery stroke, and small vessel disease).32 Evidence from each of the genetic instruments was then combined using inverse-variance weighted (IVW) meta-analysis to produce a single estimate of the causal effect of each exposure on each outcome.33
To facilitate the interpretation of results for the clinical outcomes, we also performed MR to estimate the effect of γ′ fibrinogen and total fibrinogen levels on D-dimer levels. As a product of fibrinolysis, higher levels of D-dimer result from increased formation and subsequent breakdown of fibrin clots. Positive causal effect estimates for D-dimer are therefore evidence of procoagulant activity, whereas negative causal effect estimates are evidence of anticoagulant activity.34,35
We used a Bonferroni-corrected significance threshold of P = .0042 to correct for the 12 statistical tests performed in the primary analysis.
Mendelian randomization: sensitivity analyses
In addition to the main IVW analysis, we performed several sensitivity analyses, each being robust to pleiotropic variants in certain scenarios. Sensitivity analyses included MR-Egger,36 weighted median estimator,37 weighted mode estimator,38 IVW excluding outlying instruments detected by MR-PRESSO,39 and IVW including only instruments located at the fibrinogen gene cluster. We used the global test implemented in the MR-PRESSO R package to detect heterogeneity among the genetic instruments.39 When the global test was significant (P < .05), we removed statistically significant outliers detected by MR-PRESSO (P < .05) and repeated the IVW analysis.
As an additional sensitivity analysis unrelated to pleiotropy, the Mendelian Randomization R package was used to perform IVW MR analyses corrected for LD between genetic instruments.40,41 Although genetic instruments were selected to be largely independent (r2 < .1), this method was used to determine the effects of residual LD on the primary IVW analyses.
Multivariable Mendelian randomization
To account for specific potential sources of pleiotropy, based on existing biological knowledge of the exposure, we used multivariable MR (MVMR).42 This is a regression-based approach that consists of 2 stages: (1) the β coefficients for the genetic association between the exposure and the outcome are regressed on the β coefficients for the genetic association between a third variable and the outcome, and (2) these residuals are then regressed on the β coefficients for the exposure. Because there may be an association between some of the γ′ fibrinogen and total fibrinogen instruments, we performed MVMR adjusting γ′ fibrinogen levels for total fibrinogen levels and vice versa. We also performed MVMR analyses adjusting γ′ fibrinogen levels and total fibrinogen levels for CRP levels. Like fibrinogen, CRP is part of the acute-phase response and a marker of inflammation; thus, adjusting for its levels serves to adjust for potential genetic pleiotropy related to inflammation.
Results
GWAS of γ′ fibrinogen
In the GWAS of γ′ fibrinogen levels, the only locus that was associated at genome-wide significance (P < 5 × 10−8) was the fibrinogen gene cluster itself: a total of 509 variants at the chromosome 4 locus were associated with γ′ fibrinogen levels. The most significant variant at this locus was rs7654093 with a minor allele frequency of 24%. This variant was associated with a 0.2 log(mg/dL) decrease in γ′ fibrinogen levels per copy of the minor allele (P = 3.1 × 10−418). This variant is in strong LD (r2 = .98), with 2 of the 4 variants (rs2066854 and rs2066861) from the FGG H2 haplotype that has previously been associated with γ′ fibrinogen.8 The rs7654093 variant is also tightly linked (r2 = .92) to the most significant variant (rs7681423) from a previously published GWAS of γ′ fibrinogen levels.43 Manhattan and QQ plots that summarize the γ′ fibrinogen GWAS are shown in supplemental Figures 1 and 2, available on the Blood Web site, and a regional association plot for the fibrinogen gene cluster is shown in supplemental Figure 3.
Selection of genetic instruments
After pruning the 509 genome-wide significant variants for γ′ fibrinogen levels, all of which were located at the fibrinogen gene cluster, 16 independent genetic variants remained and were used as genetic instruments for γ′ fibrinogen. After pruning the 5076 genome-wide significant variants for total fibrinogen levels, 75 independent genetic variants remained and were used as genetic instruments for total fibrinogen levels. Of these 75 variants, 16 were located at the fibrinogen gene cluster. The effects of each individual instrument on fibrinogen and γ′ fibrinogen are shown in supplemental Tables 1 and 2. Assuming complete independence of the genetic instruments, the combined variance in γ′ fibrinogen levels explained by the 16 genetic instruments for γ′ fibrinogen was 47.8% (supplemental Table 1). The single strongest genetic instrument for γ′ fibrinogen explained 19.6% of the variance. The 75 genetic instruments for total fibrinogen levels explained 4.0% of the variance of total fibrinogen levels. Each of the instruments had an F statistic >10.
Of the 16 instruments for γ′ fibrinogen levels, 14 were also associated with total fibrinogen levels (P < .05), of which 11 were associated in the same direction and 5 were associated in the opposite direction. Notably, all 16 instruments for γ′ fibrinogen had a more significant P value in the GWAS of γ′ fibrinogen levels than in the GWAS of total fibrinogen levels, despite the smaller sample size of the GWAS of γ′ fibrinogen levels. This provides evidence for their suitability as γ′ fibrinogen instruments, because it suggests that the 16 genetic variants selected for γ′ fibrinogen primarily regulate the differential expression of fibrinogen isoforms as opposed to primarily affecting total fibrinogen levels. Of the 75 instruments for total fibrinogen levels, 32 were also associated with γ′ fibrinogen levels (P < .05), of which 15 were associated in the same direction and 17 were associated in the opposite direction.
Supplemental Table 3 shows the degree of overlap between instruments selected for γ′ fibrinogen and the well-studied FGG H2 haplotype. Only one of the instruments, rs2066854, was in strong LD (r2 = .96) with rs2066861, a genetic variant that tags the FGG H2 haplotype. The remaining instruments largely provided information that was independent of the FGG H2 haplotype (LD, r2 < .1).
MR considering γ′ fibrinogen as the exposure
Odds ratios (ORs) and 95% confidence intervals (CIs) from IVW MR for the association of genetically determined γ′ fibrinogen levels with VTE, any ischemic stroke, and stroke subtypes are shown in Figure 1A. Forest plots that show the causal effect estimate on each clinical outcome produced using each individual genetic instrument for γ′ fibrinogen levels are shown in supplemental Figure 4.
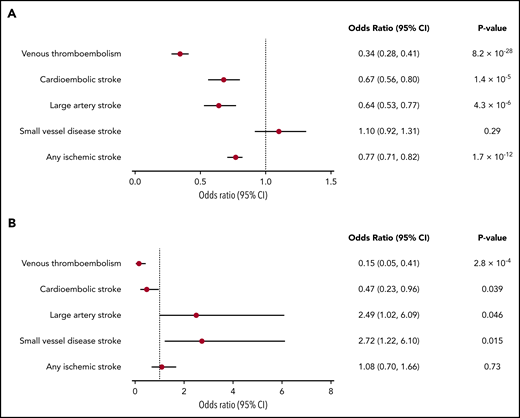
Forest plots of the association of genetically determined γ′ fibrinogen and total fibrinogen levels with VTE and ischemic stroke subtypes. (A) γ′ fibrinogen levels; (B) total fibrinogen levels.
Forest plots of the association of genetically determined γ′ fibrinogen and total fibrinogen levels with VTE and ischemic stroke subtypes. (A) γ′ fibrinogen levels; (B) total fibrinogen levels.
Genetically determined γ′ fibrinogen levels were inversely associated with the risk of VTE, with an ORIVW of 0.34 per log(mg/dL) difference in γ′ fibrinogen levels (95% CI: 0.28-0.41; PIVW = 8.2 × 10−28), as well as with cardioembolic stroke (ORIVW = 0.67; 95% CI: 0.56-0.80; PIVW = 1.4 × 10−5), large artery stroke (ORIVW = 0.64; 95% CI: 0.53-0.77; PIVW = 4.3 × 10−6), and any ischemic stroke (ORIVW = 0.77; 95% CI: 0.71-0.82; PIVW = 1.7 × 10−12). The associations had consistent effect size and directions across sensitivity analyses (supplemental Figure 5). Genetically determined γ′ fibrinogen levels were not associated with small vessel disease (ORIVW = 1.10; 95% CI: 0.92-1.31; PIVW = .29). As shown in supplemental Table 4A, none of the β coefficients for the MR-Egger intercept differed significantly from 0 (P < .05).
There is evidence that γ′ fibrinogen affects clot structure and therefore may differentially affect the 2 components of VTE: deep vein thrombosis and pulmonary embolism. We therefore used a publicly available phenome-wide association study in the UK Biobank (http://pheweb.sph.umich.edu) to examine the association of the strongest genetic instrument for γ′ fibrinogen, rs2066854, with deep vein thrombosis and pulmonary embolism separately. As shown in supplemental Table 5, rs2066854 was associated with deep vein thrombosis and pulmonary embolism in the same direction and to a similar degree.
MR considering total fibrinogen as the exposure
Effect estimates of genetically determined total fibrinogen levels on VTE, any ischemic stroke, and ischemic stroke subtypes, generated using the IVW approach, are shown in Figure 1B. Forest plots that show the causal effect estimate on each clinical outcome produced using each individual genetic instrument for total fibrinogen levels are shown in supplemental Figure 6.
Genetically determined total fibrinogen levels were inversely associated with the risk of VTE, with an ORIVW of 0.15 per log(g/L) difference in total fibrinogen levels (95% CI: 0.05-0.41; PIVW = 2.8 × 10−4). This association was consistent across sensitivity analyses (supplemental Figure 7). There were also a number of suggestive associations that were not significant according to our Bonferroni-corrected P value threshold, including a possible inverse association with cardioembolic stroke (ORIVW = 0.47; 95% CI: 0.23-0.96; PIVW = .039) and possible positive associations with small vessel disease (ORIVW = 2.72; 95% CI: 1.22-6.10; PIVW = .015) and large artery stroke (ORIVW = 2.49; 95% CI: 1.02-6.09; PIVW = .046). Sensitivity analyses were consistent with a positive effect direction for small vessel disease but did not consistently show a positive effect direction for large artery stroke (supplemental Figure 7). Finally, genetically determined total fibrinogen levels were not clearly associated with any ischemic stroke (Figure 1B; supplemental Figure 7). In our primary analysis, none of the β coefficients for the MR-Egger intercept differed significantly from 0 (supplemental Table 4B). However, when performing MR-Egger after removing the outlying instrument identified by MR-PRESSO, a significant (P = .003) nonzero MR-Egger intercept was found for the association between total fibrinogen and VTE (supplemental Table 4C).
Multivariable Mendelian randomization
Effect estimates for γ′ fibrinogen levels adjusted for total fibrinogen levels and CRP levels using MVMR are presented in supplemental Figure 5 alongside the other sensitivity analyses. When adjusting the association between γ′ fibrinogen levels and the clinical outcomes for total fibrinogen levels, there was some evidence (P = .033) of an indirect effect through total fibrinogen levels on VTE (supplemental Table 6A). However, adjustment for total fibrinogen resulted in a similar OR, which was 0.34 (95% CI: 0.28-0.41) before adjustment and 0.38 (95% CI: 0.31-0.46) after adjustment. There was no evidence (P < .05) of indirect effects through total fibrinogen levels for any other considered clinical outcome (supplemental Table 6A).
Effect estimates for total fibrinogen levels adjusted for γ′ fibrinogen levels and CRP levels using MVMR are presented in supplemental Figure 7 alongside the other sensitivity analyses (supplemental Table 6B). The estimate for the effect of total fibrinogen levels on VTE was 0.15 (95% CI: 0.05-0.41) before adjustment for γ′ fibrinogen levels and 0.38 (95% CI: 0.16-0.88) after adjustment. Similarly, the estimate of the effect on cardioembolic stroke was 0.47 (95% CI: 0.23-0.96) before adjustment and 0.62 (95% CI: 0.30-1.28) after adjustment. The P values for these indirect effects through γ′ fibrinogen levels were highly significant (4.7 × 10−10 for VTE and .0046 for cardioembolic stroke). In contrast, the estimate of the effect of total fibrinogen levels on any ischemic stroke was 1.08 (95% CI: 0.70-1.66) before adjustment for γ′ fibrinogen levels and 1.37 (95% CI: 0.92-2.06) after adjustment, and the estimate for large artery stroke was 2.49 (95% CI: 1.02-6.09) before adjustment and 4.11 (95% CI: 1.70-9.90) after adjustment.
As shown in supplemental Table 6, there was no evidence (P < .05) of any indirect effects on the clinical outcomes through CRP levels except for the association between γ′ fibrinogen levels and large artery stroke (P = .0016). However, adjustment of this association for CRP levels had little impact on the association estimate and was 0.64 (95% CI: 0.53-0.77) before adjustment and 0.65 (95% CI: 0.57-0.75) after adjustment.
MR considering D-dimer as the outcome
The association of genetically determined γ′ fibrinogen and total fibrinogen levels with D-dimer levels is shown in supplemental Figure 8. Genetically determined γ′ fibrinogen levels were inversely associated with D-dimer levels, with an effect size of −0.33 log(ng/dL) per log(mg/dL) difference in γ′ fibrinogen levels (95% CI: −0.26 to −0.40; P = 1.37 × 10−19). In contrast, the association of genetically determined total fibrinogen levels with D-dimer levels was positive, albeit not significant after Bonferroni correction, with an effect size of 0.47 log(ng/dL) per log(g/L) difference in total fibrinogen levels (95% CI: 0.09-0.86; P = .015). Effect directions were consistent across sensitivity analyses.
Discussion
In the current MR study, we examined the effects of genetically determined γ′ fibrinogen levels and total fibrinogen levels on VTE and ischemic stroke subtypes. Higher genetically determined γ′ fibrinogen levels were associated with a lower risk of VTE, any ischemic stroke, cardioembolic stroke, and large artery stroke. Higher genetically determined total fibrinogen levels were associated with a lower risk of VTE, and there were indications that they may also be associated with a higher risk of small vessel disease. MR-Egger analyses did not provide any evidence of directional pleiotropy, except when removing the outlying instrument identified by MR-PRESSO for total fibrinogen and VTE.44
Although there is evidence that γ′ fibrinogen modifies fibrin clot structure and stability,45,46 if this were the main driver of the inverse association with VTE, we would expect to observe differential associations with the 2 components incorporated in the VTE outcome: deep vein thrombosis and pulmonary embolism. However, our analyses in the UK Biobank suggest that the association of γ′ fibrinogen levels is consistent across these 2 components of VTE. The γ′ fibrinogen protein has several antithrombotic properties that may offer an alternative explanation for the observed inverse association of its genetically determined levels with VTE and certain ischemic stroke subtypes, namely cardioembolic and large artery stroke. First, γ′ fibrinogen binds to thrombin with high affinity, reducing thrombin’s ability to transform fibrinogen into a fibrin and thrombin’s ability to activate platelets.47,48 Second, although fibrinogen plays an important role in platelet aggregation, γ′ fibrinogen specifically does not. Because of alternative splicing, γ′ fibrinogen has a different carboxyl-terminal region, whose mechanism reduces platelet binding.49 Third, γ′ fibrinogen may inhibit factor V and factor XIII activation.6,50 Nevertheless, γ′ fibrinogen also has a prothrombotic property: clots comprised of fibrin derived from γ′ fibrinogen have an altered structure that increases their resistance to fibrinolysis.51 Overall, the MR results suggest that despite this prothrombotic property, the net effect of γ′ fibrinogen is antithrombotic. D-dimer is a byproduct of fibrinolysis, and high rates of fibrinolysis usually occur in response to high rates of fibrin clot formation. The significant inverse association between genetically determined γ′ fibrinogen levels and D-dimer levels therefore suggests that the protective effect of γ′ fibrinogen levels on VTE, any ischemic stroke, cardioembolic stroke, and large artery stroke may be driven by reduced rates of fibrin clot formation. If so, this is likely attributable to γ′ fibrinogen’s inhibitory binding of thrombin. The differential effect of γ′ fibrinogen on different stroke subtypes may reflect differences in the etiologies of the subtypes. Thrombosis plays a prominent role in both cardioembolic and large artery stroke, but a role of thrombosis in small vessel disease is less apparent.31 Correspondingly, levels of γ′ fibrinogen appear to be primarily associated with a lower risk of cardioembolic and large artery stroke and not small vessel disease.
Our results are in line with previous studies that linked the γ′ fibrinogen decreasing FGG H2 haplotype to an increased risk of VTE.8 Our results expand on the prior studies by including 16 genetic instruments for γ′ fibrinogen and showing an association between genetically determined γ′ fibrinogen levels and VTE that extends beyond the FGG H2 haplotype. Only 1 of the 16 instruments is tightly linked to the FGG H2 haplotype. This is important, because causal inference based on this single genetic instrument may not be valid if the instrument has any pleiotropic effects on VTE that are not mediated by γ′ fibrinogen. By using 16 genetic instruments, we were able to perform a range of sensitivity analyses to explore the role pleiotropy, each of which requires the use of more than 1 genetic instrument.
Based on fibrinogen’s role in clot formation and platelet aggregation, observational studies, and a recent MR study on the effect of total fibrinogen on coronary heart disease,52 we hypothesized that increased levels of total fibrinogen would be associated with increased risk of VTE and ischemic stroke. Indeed, the direction of the association between genetically determined levels of total fibrinogen and D-dimer levels was positive, which is consistent with higher total fibrinogen levels leading to higher rates of fibrin clot formation. For small vessel disease and large artery stroke we did observe a positive association, but contrary to our hypothesis, increased genetically determined total fibrinogen levels were associated with a lower risk of VTE. Our MVMR analyses suggests that the protective association of total fibrinogen levels with VTE may at least be in part explained by violations of MR assumptions in the form of indirect effects of the instrumental variables through γ′ fibrinogen levels. Once γ′ fibrinogen levels were adjusted for, attenuation of the effect estimates of total fibrinogen on VTE suggest that pleiotropy through γ′ fibrinogen levels may be biasing the estimates for total fibrinogen levels on these outcomes away from the null. In contrast, the effect estimates of total fibrinogen on any ischemic stroke and large artery stroke moved away from the null on adjustment for γ′ fibrinogen levels, suggesting γ′ fibrinogen is biasing effect estimates for total fibrinogen levels on these outcomes toward the null.
This study has several limitations. There is some overlap between the participants included in the GWAS of the exposures (γ′ fibrinogen and total fibrinogen) and the participants included in the clinical outcomes. This can potentially bias the results, although the extent of the bias depends on the degree of sample overlap, which was generally low.53 Furthermore, MR effect estimates are only equivalent to causal effects when the assumptions of MR are met. Pleiotropic effects of genetic instruments on the outcome that are not mediated by the exposure represent a particularly important violation of MR assumptions and can lead to biased effect estimates.54 There is no test that can robustly rule out the presence of pleiotropic effects among the genetic instruments, so to minimize the impact of pleiotropy we used a wide variety of sensitivity analyses, each with their own assumptions about the pleiotropic effects of the instruments.36-39 Without knowing the nature of pleiotropic genetic instruments and the extent to which they are violating this assumption, we cannot know which sensitivity analysis is most accurate. However, a broad consensus across sensitivity analyses support that bias from pleiotropy is minimal, and this was the case for most reported associations. Our algorithm for selecting genetic instruments allowed for multiple variants to be selected at any given locus, as long as these were not in LD with each other (r2 < .1). As a result, we used multiple variants at the fibrinogen gene cluster as genetic instruments. To control for potential bias related to any residual LD (r2 < .1) among these variants, we performed a separate sensitivity analysis designed to account for LD, and this did not greatly change our results. Finally, the GWAS of γ′ fibrinogen levels were limited because of smaller sample size than the GWAS of fibrinogen levels. As a result, the only genome-wide significant locus was the fibrinogen gene cluster. However, this was the largest available GWAS of γ′ fibrinogen levels available at the time of the analyses.
In summary, we found that higher genetically determined γ′ fibrinogen levels were associated with a lower risk of VTE, any ischemic stroke, cardioembolic stroke, and large artery stroke. These results are in line with documented antithrombotic effects of γ′ fibrinogen and may be indicative of a causal effect of γ′ fibrinogen on these outcomes. These results complement prior studies that have used MR to identify potentially causal associations of other coagulation proteins with VTE and stroke, including factor VII, factor VIII, factor XI, and von Willebrand factor.55-57
Summary statistics of genome-wide association studies of total fibrinogen levels are available on dbGaP (phs000930), and we will place summary statistics for γ prime fibrinogen in the same location. Summary statistics for ischemic stroke subtypes are publicly available at https://www.megastroke.org/download.html. Summary statistics for venous thromboembolism are available from the INVENT consortium upon request.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the staff and participants of the ARIC study for their important contributions.
A.S.W., M.F., N.L.S., A.C.M., and P.S.d.V. were supported by National Institutes of Health (NIH), National Heart, Lung, and Blood Institute (NHLBI) grant R01HL141291. N.L.S., A.C.M., B.M., and P.S.d.V. were supported by NIH/NHLBI grant R01HL139553. P.S.d.V. was additionally supported by American Heart Association (AHA) grant 18CDA34110116. N.P. and W.T. were supported by NIH/NHLBI grant R01HL59367. D.G. was supported by the Wellcome Trust 4i Programme (203928/Z/16/Z) and British Heart Foundation Centre of Research Excellence (RE/18/4/34215) at Imperial College London. The CHARGE Consortium Hemostasis Working Group is partially supported by NIH/NHLBI grant R01HL134894, and infrastructure for the CHARGE Consortium is supported, in part, by NIH/NHLBI grant R01HL105756. The Atherosclerosis Risk in Communities (ARIC) study has been funded in whole, or in part, with federal funds from the NIH/NHLBI, Department of Health and Human Services (contracts HHSN268201700001I, HHSN268201700002I, HHSN268201700003I, HHSN268201700004I, and HHSN268201700005I); Grants R01HL087641, R01HL059367, and R01HL086694; NIH/National Human Genome Research Institute (NHGRI) contract U01HG004402; and NIH contract HHSN268200625226C. Infrastructure was partly supported by grant UL1RR025005, a component of the NIH and NIH Roadmap for Medical Research. Measurement of γ′ fibrinogen in the ARIC study was supported by NIH/NHLBI grant R01HL59367.
Authorship
Contribution: P.S.d.V. and A.D. designed the research; J.M., D.G., and N.P. performed statistical analyses; S.L., W.T., M.F., S.D., M.D. contributed vital new data; J.M., D.G., M.A.L., A.S.W., M.P.M.d.M., C.K.W.-C., B.M., E.B., N.L.S., A.C.M., A.D., and P.S.d.V. contributed to interpretation of the results; J.M., D.G., A.D., and P.S.d.V. wrote the paper; and all authors critically revised the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Complete lists of the members of the CHARGE Inflammation Working Group, the INVENT Consortium, and the MEGASTROKE consortium of the International Stroke Genetics Consortium (ISGC) appear in the supplemental appendix.
Correspondence: Paul S. de Vries, Human Genetics Center, Department of Epidemiology, Human Genetics, and Environmental Sciences, School of Public Health, The University of Texas Health Science Center at Houston, 1200 Pressler St, Suite E-429, Houston, TX 77030; e-mail: paul.s.devries@uth.tmc.edu.
