

In this issue of Blood, 1 describe studies in a murine model of congenital erythropoietic porphyria (CEP). They treated CEP mice for 26 weeks with deferiprone (an iron chelator) and report that it reversed their skin photosensitivity and hemolytic anemia. The murine model closely mirrors human disease, and thus, these results are both provocative and important.
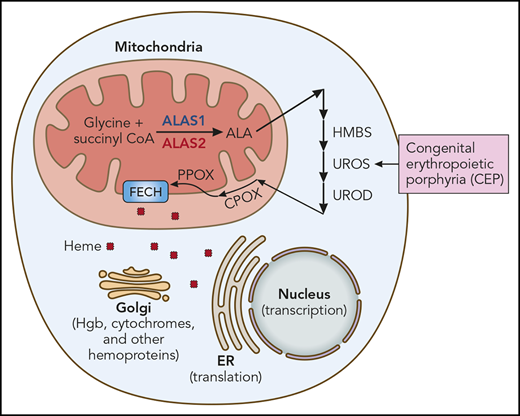
Heme synthesis and use. The heme synthetic pathway and heme’s intracellular trafficking are diagrammed. The first and rate-limiting step is catalyzed by ALAS. ALAS2 is the erythroid-specific ALAS isoform (red). The presence of an iron responsive element (IRE) in the 5′ untranslated region of ALAS2 mRNA renders it exquisitely sensitive to iron. All other cells, including hepatocytes, contain ALAS1 (blue). CEP results from mutation of UROS, the fourth enzyme of the heme synthetic pathway. Other porphyrias result from mutations of other heme synthetic pathway enzymes, eg, HMBS (hydroxymethylbilane synthase; acute intermittent porphyria), UROD (uroporphyrinogen decarboxylase; porphyria cutanea tarda), CPOX (coproporphyrinogen oxidase; hereditary coproporphyria), PPOX (protoporphyrinogen oxidase; variegate porphyria), and FECH (ferrochelatase; erythropoietic protoporphyria). Iron chelation ameliorates CEP by reducing ALAS2 translation, thus decreasing heme synthesis and the buildup of pathway intermediates proximal to UROS and their phototoxic metabolites. ER, endoplasmic reticulum; Hgb, hemoglobin. Professional illustration by Patrick Lane, ScEYEnce Studios.
Heme synthesis and use. The heme synthetic pathway and heme’s intracellular trafficking are diagrammed. The first and rate-limiting step is catalyzed by ALAS. ALAS2 is the erythroid-specific ALAS isoform (red). The presence of an iron responsive element (IRE) in the 5′ untranslated region of ALAS2 mRNA renders it exquisitely sensitive to iron. All other cells, including hepatocytes, contain ALAS1 (blue). CEP results from mutation of UROS, the fourth enzyme of the heme synthetic pathway. Other porphyrias result from mutations of other heme synthetic pathway enzymes, eg, HMBS (hydroxymethylbilane synthase; acute intermittent porphyria), UROD (uroporphyrinogen decarboxylase; porphyria cutanea tarda), CPOX (coproporphyrinogen oxidase; hereditary coproporphyria), PPOX (protoporphyrinogen oxidase; variegate porphyria), and FECH (ferrochelatase; erythropoietic protoporphyria). Iron chelation ameliorates CEP by reducing ALAS2 translation, thus decreasing heme synthesis and the buildup of pathway intermediates proximal to UROS and their phototoxic metabolites. ER, endoplasmic reticulum; Hgb, hemoglobin. Professional illustration by Patrick Lane, ScEYEnce Studios.
CEP is an autosomal recessive disorder that is caused by mutation of uroporphyrinogen synthase III (UROS), the fourth enzyme of the heme synthetic pathway (see figure). Reduced UROS activity leads to the accumulation of phototoxic metabolites in late erythroid precursors in marrow, reticulocytes, and circulating red blood cells. This results in bullous lesions in skin exposed to sunlight (or UV light), skin scarring and retraction, hand and face mutilation, erythrodontia (brown discoloration of teeth), corneal scarring, and hemolysis. The quantity of residual UROS activity determines clinical severity. Although the biochemistry of heme synthesis and the pathogenesis of CEP are well understood, CEP therapy is limited to protective clothing, strict sun avoidance, splenectomy, and other supportive measures.2 That some patients remit after marrow transplantation3 confirms that the pathology results from aberrant heme synthesis in erythroid cells, and for this reason, CEP is termed an erythropoietic (not hepatic) porphyria.
In rare disorders such as CEP, rigorous clinical trials are difficult, and therapeutic efficacy is generally demonstrated through a series of case studies. Egan et al4 previously described a CEP patient whose devastating skin lesions and hemolysis remitted for >2 years when she was treated with deferasirox (also an iron chelator). Mirmiran et al5 previously described a CEP patient whose photosensitivity and hemolysis remitted with frequent phlebotomy. The uniform and dramatic clinical response to iron restriction of the 15 CEP mice studied by Blouin et al and these 2 CEP patients4,5 argues that restricting iron, via either chelation or phlebotomy, should be the front-line therapy for this devastating disorder. In the CEP mice and patients, iron restriction was efficacious without inducing iron deficiency anemia.
Porphyrias broadly result from the mutation of enzymes that catalyze heme synthesis. Heme is needed for hemoglobin, cytochromes, myoglobin, catalases, heme peroxidase, and endothelial nitric oxide synthase. Heme also regulates transcription (via Bach1) and translation (via HRI) and is synthesized by all cells. However, the erythron and liver produce massive amounts of heme for hemoglobin and cytochromes, respectively, and hence are most sensitive to mutations that block this pathway.
The heme synthetic pathway (see figure) is unusual. It begins with glycine (an amino acid) and succinyl CoA (a tricarboxylic acid cycle intermediate) and thus senses amino acid and energy availability. The first and rate-limiting step, the conversion of the initial substrates to δ-aminolevulinic acid (ALA), is catalyzed by aminolevulinic acid synthase (ALAS) and takes place in mitochondria. The next 4 steps are cytoplasmic (including UROS), while the final steps of heme synthesis occur in the mitochondria. Heme then traffics to all cellular compartments to complete its multiple missions. Decreasing ALAS should limit substrate entrance into the heme synthetic pathway and mitigate porphyria.
Importantly, the regulation of heme synthesis differs in the erythron and liver. The ALAS isoform, ALAS2 (red in figure), is the first and rate-limiting step of erythroid heme synthesis, while ALAS1 (blue in figure) drives heme synthesis in all other cells, including liver. These enzymes are encoded by genes on different chromosomes (X chromosome and chromosome 3, respectively), share little amino acid homology, and are regulated differently.
Although transcriptionally and translationally regulated by iron, ALAS1 is predominantly regulated via negative feedback from heme. When heme is plentiful, the transfer of ALAS1 from the cytoplasm to the mitochondria (its site of activity) is impaired. For this reason, hemin (heme) is an effective therapy for the hepatic porphyrias, such as acute intermittent porphyria (AIP). Similarly, a newly approved drug, givosiran, a small interfering RNA that inhibits ALAS1 expression, improves AIP symptoms.6 ALAS2 is not regulated by heme, and thus hemin therapy is not efficacious in CEP. Givosiran targets a sequence unique to ALAS1 and thus cannot ameliorate CEP. Also, givosiran delivery to hepatocytes is via the asilaoglycoprotein receptor and liver-specific, so givosiran cannot enter erythroid cells. Importantly, however, ALAS2 messenger RNA (mRNA), by virtue of its 5′ IRE, is exquisitely sensitive to iron. Iron stabilizes ALAS2 mRNA, leading to more protein production and more heme synthesis. Iron restriction reverses this. Ancillary studies by Blouin et al and those by Egan et al4 and Mirmiran et al5 show that iron restriction limits ALAS2 protein production and activity and confirm the molecular mechanism. By extension, other agents that target ALAS2 should also be useful therapies.
Theoretically, iron restriction could reduce liver, as well as erythroid, heme production, perhaps by effects on iron sulfur complexes or ferrochelatase (FECH) function, to result in less heme, more ALAS1, increased toxic metabolites in liver, and hepatic damage. However, it is encouraging that this was not observed in the murine CEP model.1
The work of Blouin et al, by studying a large cohort of CEP mice whose clinical phenotype closely resembles CEP patients, is impactful and could change therapy for patients with this rare disorder. This work further emphasizes the importance of animal models that clinically and pathophysiologically mirror human disease.
Conflict-of-interest disclosure: The author declares no competing financial interests.
