

Abstract
Activated B-cell (ABC)-diffuse large B-cell lymphomas (DLBCLs) are clinically aggressive and phenotypically complex malignancies, whose transformation mechanisms remain unclear. Partially differentiated antigen-secreting cells (plasmablasts) have long been regarded as cells-of-origin for these tumors, despite lack of definitive experimental evidence. Recent DLBCL reclassification based on mutational landscapes identified MCD/C5 tumors as specific ABC-DLBCLs with unfavorable clinical outcome, activating mutations in the signaling adaptors MYD88 and CD79B, and immune evasion through mutation of antigen-presenting genes. MCD/C5s manifest prominent extranodal dissemination and similarities with primary extranodal lymphomas (PENLs). In this regard, recent studies on TBL1XR1, a gene recurrently mutated in MCD/C5s and PENLs, suggest that aberrant memory B cells (MBs), and not plasmablasts, are the true cells-of-origin for these tumors. Moreover, transcriptional and phenotypic profiling suggests that MCD/C5s, as a class, represent bona fide MB tumors. Based on emerging findings we propose herein a generalized stepwise model for MCD/C5 and PENLs pathogenesis, whereby acquisition of founder mutations in activated B cells favors the development of aberrant MBs prone to avoid plasmacytic differentiation on recall and undergo systemic dissemination. Cyclic reactivation of these MBs through persistent antigen exposure favors their clonal expansion and accumulation of mutations, which further facilitate their activation. As a result, MB-like clonal precursors become trapped in an oscillatory state of semipermanent activation and phenotypic sway that facilitates ulterior transformation and accounts for the extranodal clinical presentation and biology of these tumors. In addition, we discuss diagnostic and therapeutic implications of a MB cell-of-origin for these lymphomas.
Introduction
Diffuse large B-cell lymphomas (DLBCLs) arise from the highly dynamic adaptive immune system, in which B cells transit through various phenotypic/transitional states before differentiating into antibody-secreting cells.1 Resolving the trajectories through which DLBCLs emerge is important because it could provide the basis for earlier detection of malignant precursor lesions and inform rationally designed therapeutic approaches. The first molecular classification of DLBCLs grouped tumors into general categories (germinal center B-cell [GCB]-derived, activated B-cell [ABC]-derived or unclassified) by comparing their transcriptomes to that of broad normal B-cell populations.2 Among these, the cell-of-origin (COO) and transformation mechanisms of ABC-DLBCLs remain generally obscure, despite the fact that these are among the most aggressive and incurable lymphomas.3
Recent studies4-6 reclassified DLBCLs based on constellations of genetic lesions, creating an opportunity to refine our understanding of ABC-DLBCL lymphomagenesis. In particular, 2 studies5,6 described a genetically defined ABC-DLBCL subtype (MCD or Cluster 5 [C5]) that features hallmark activating mutations in MYD88 and CD79B and loss-of-function mutations in the poorly characterized gene TBL1XR1.7 MCD/C5s are highly aggressive tumors with unfavorable clinical outcome, frequent extranodal dissemination, and immune surveillance evasion.4,6 Indeed, MCD/C5 genomic landscapes strikingly resemble that of primary extranodal lymphomas (PENLs).8,9 PENLs reflect an extreme DLBCL presentation, in that they occupy extranodal and even immune-privileged sites, such as the central nervous system (CNS), vitreo-retina, or testes, without prior growth in lymphoid organs.10 Curiously, neither GCBs nor plasma cells (PCs) normally home to these tissues, raising questions about the origin of these tumors within the complex milieu of B-cell populations.
Despite their elusive origins, ABC-DLBCLs are thought to derive from B cells that have transited through a germinal center (GC) reaction11 (Table 1), because B-cell receptors (BCRs) in these tumors show evidence of somatic hypermutation (SHM), a process generally believed to be GCB specific.1 The fact that these tumors frequently present an immunoblastic histology12 and express the transcription factor (TF) IRF4,13 required for PC differentiation and survival,14,15 led to a widely accepted notion that ABC-DLBCLs originate from plasmablasts (PBs; PC precursor cells). However, recent mass cytometry profiling of MCD/C5 clinical specimens suggests that, instead, these more closely recapitulate a memory B cell (MB)-like phenotype.7 Indeed, MCD/C5 transcriptional profiles were found to be more closely related to normal precursor MB populations and depleted for PB16 or PC4 signatures. Furthermore, modeling TBL1XR1 mutations in mice led to the unexpected discovery that MCD/C5s might develop through cyclic and repetitive reactivation of MB subpopulations.7 These data suggest that MCD/C5s are actually tumors of highly malignant and aggressive MBs. This perspective will address the implications of these novel findings and propose a generalized model for MCD/C5 and PENL pathogenesis, providing a rationale for the distinctive clinical features of these tumors and highlighting potential new therapeutic approaches. Of note, recent studies on N1 DLBCLs4,16 (another ABC-DLBCL subclass) or ABC-DLBCLs as a whole16 also pointed to MBs as their closest normal counterpart, suggesting that aspects of the models herein could be generalized to ABC-DLBCLs beyond MCD/C5s.
Glossary of terms related to B-cell immune responses
| Term | Definition |
|---|---|
| AID (AICDA) | Enzyme responsible for driving the somatic hypermutation and class switch recombination mechanisms in activated B cells, both of which contribute to B-cell receptor diversification. |
| Affinity maturation | Process by which GCBs develop B-cell receptors with increased antigen affinity through repeated rounds of diversification, competitive selection, and clonal expansion. |
| Anergy | Condition in which mature B cells persist in periphery but are poorly responsive to antigen, responsible for silencing self-reactive B cells. Anergy loss contributes to autoimmune disorders. |
| Antigen or molecular mimicry | Phenomena in which sequence similarities between foreign and self-antigens are sufficient to result in the cross-activation of auto-reactive B cells by pathogen-derived antigens. |
| Antigen presentation | Surveillance process, essential for T-cell activation, in which T cells screen short peptide antigens displayed on the surface of other cells. |
| B-cell receptor | Membrane-bound immunoglobulin-type receptor, acquired early during B-cell development, that recognizes and binds specific antigens causing activation of mature B cells. |
| B-cell receptor or antibody repertoire | Collection of B-cell receptors/immunoglobulin sequences expressed by a given population of B cells. |
| Class switch recombination/isotype switching | DNA recombination process by which the B-cell receptor constant portion is exchanged in mature activated B cell, generating functional diversity while maintaining antigen specificity. |
| Clonal precursor cells | Genetically distinct subpopulations of B cells thought to clonally derive from a single founding cell which, following acquisition of one or more somatic mutations, gained a disproportionate proliferative advantage over other mature B-cell populations. |
| Follicular dendritic cells | Nonhematopoietic stromal cells in B-cell follicles and GCs that retain antigens at their cell surface in a manner crucial to the selection of B cells expressing high-affinity antigen receptors. |
| Germinal center reaction | Transient immune structures formed in lymphoid organs, in which activated B cells proliferate, mutate their B-cell receptors, and differentiate to generate high-affinity antibodies and immunological memory. |
| Immune synapse | Specialized cell–cell junction between T cells and antigen-presenting cells, such as GCBs, that allows focal mutual interaction via soluble and membrane-bound factors. |
| Off-target mutations | Also known as aberrant somatic hypermutation (aSHM). Sporadic mutations introduced by AID in loci beyond the B-cell receptor, as a byproduct of the somatic hypermutation mechanism in activated B cells. |
| Recall response | Adaptive immune reaction mounted by memory B cells on re-encounter with identical or closely related antigenic challenges. These secondary responses tend to be faster and enhanced compared with original/primary immune responses. |
| Self-reactivity | Recognition of autologous antigens by a B-cell receptor, potentially capable of evoking a pathogenic immune response by the host. |
| Somatic hypermutation | Mechanism of B-cell receptor sequence diversification through locus-directed DNA mutagenesis, catalyzed by the enzyme AID in activated mature B cells. |
| T-follicular helper cells | Specialized subset of CD4+ T-cells essential for GC formation and maintenance, affinity maturation, and development of most high-affinity antibodies and memory B cells. |
| Term | Definition |
|---|---|
| AID (AICDA) | Enzyme responsible for driving the somatic hypermutation and class switch recombination mechanisms in activated B cells, both of which contribute to B-cell receptor diversification. |
| Affinity maturation | Process by which GCBs develop B-cell receptors with increased antigen affinity through repeated rounds of diversification, competitive selection, and clonal expansion. |
| Anergy | Condition in which mature B cells persist in periphery but are poorly responsive to antigen, responsible for silencing self-reactive B cells. Anergy loss contributes to autoimmune disorders. |
| Antigen or molecular mimicry | Phenomena in which sequence similarities between foreign and self-antigens are sufficient to result in the cross-activation of auto-reactive B cells by pathogen-derived antigens. |
| Antigen presentation | Surveillance process, essential for T-cell activation, in which T cells screen short peptide antigens displayed on the surface of other cells. |
| B-cell receptor | Membrane-bound immunoglobulin-type receptor, acquired early during B-cell development, that recognizes and binds specific antigens causing activation of mature B cells. |
| B-cell receptor or antibody repertoire | Collection of B-cell receptors/immunoglobulin sequences expressed by a given population of B cells. |
| Class switch recombination/isotype switching | DNA recombination process by which the B-cell receptor constant portion is exchanged in mature activated B cell, generating functional diversity while maintaining antigen specificity. |
| Clonal precursor cells | Genetically distinct subpopulations of B cells thought to clonally derive from a single founding cell which, following acquisition of one or more somatic mutations, gained a disproportionate proliferative advantage over other mature B-cell populations. |
| Follicular dendritic cells | Nonhematopoietic stromal cells in B-cell follicles and GCs that retain antigens at their cell surface in a manner crucial to the selection of B cells expressing high-affinity antigen receptors. |
| Germinal center reaction | Transient immune structures formed in lymphoid organs, in which activated B cells proliferate, mutate their B-cell receptors, and differentiate to generate high-affinity antibodies and immunological memory. |
| Immune synapse | Specialized cell–cell junction between T cells and antigen-presenting cells, such as GCBs, that allows focal mutual interaction via soluble and membrane-bound factors. |
| Off-target mutations | Also known as aberrant somatic hypermutation (aSHM). Sporadic mutations introduced by AID in loci beyond the B-cell receptor, as a byproduct of the somatic hypermutation mechanism in activated B cells. |
| Recall response | Adaptive immune reaction mounted by memory B cells on re-encounter with identical or closely related antigenic challenges. These secondary responses tend to be faster and enhanced compared with original/primary immune responses. |
| Self-reactivity | Recognition of autologous antigens by a B-cell receptor, potentially capable of evoking a pathogenic immune response by the host. |
| Somatic hypermutation | Mechanism of B-cell receptor sequence diversification through locus-directed DNA mutagenesis, catalyzed by the enzyme AID in activated mature B cells. |
| T-follicular helper cells | Specialized subset of CD4+ T-cells essential for GC formation and maintenance, affinity maturation, and development of most high-affinity antibodies and memory B cells. |
Features that define MBs as lymphoma cells-of-origin
MBs are phenotypically and functionally diverse cells that provide durable responsiveness to immunologic challenges, enabling faster and enhanced immunity on re-encounter with the same or closely related antigens.17 MBs are long-lived and can persist for decades,18 retain self-renewal potential when activated,19 and share gene expression programs with long-term hematopoietic stem cells.20 Conversely, most PCs generated during primary immune responses extinguish shortly after resolution, with only a small fraction persisting as long-lived terminally differentiated cells that home to the bone marrow (BM).21 These traits point to MBs as more suitable vessels for lengthy stepwise lymphomagenesis mechanisms (Figure 1).
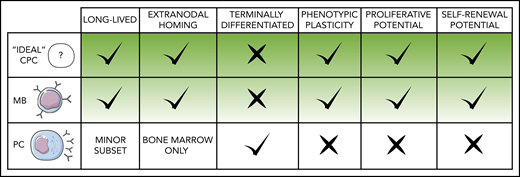
Features that define MBs as lymphoma COO. Predicted characteristics of MCD/C5 and PENLs clonal precursor cells compared with normal MB and PC defining traits. Given the heterogeneous nature of these populations, particular subclasses of MBs and PCs may deviate to some extent from the norm represented here.
Features that define MBs as lymphoma COO. Predicted characteristics of MCD/C5 and PENLs clonal precursor cells compared with normal MB and PC defining traits. Given the heterogeneous nature of these populations, particular subclasses of MBs and PCs may deviate to some extent from the norm represented here.
Despite their basal quiescence, MB signaling pathways22 and epigenetic patterning23 support their prompt reactivation in response to immune signals at lower input thresholds than naive B cells (NBs). On reactivation, MB plasticity allows them to undergo terminal plasmacytic differentiation or seed new GC reactions.17 Mechanisms determining alternative cell fates remain largely unknown, but most MBs are biased toward PC differentiation,24 and GC re-entry is infrequent.25 The limited subset of MBs that do repopulate new GCs typically maintain an immunoglobulin M (IgM)-type BCR19,26,27 and are thought to arise early during the primary GC.28 Under normal circumstances, GC re-entry permits further specification of antibody repertoires by having antigen-experienced cells undergo new rounds of affinity maturation, but this mechanism can be hijacked and exacerbated by MCD/C5 canonical mutations to favor transformation.7
Beyond these points, MBs intrinsic capacity to disseminate throughout the body further supports them as precursors for PENLs. Unlike long-lived PCs (LLPCs), largely confined to the BM,21 MBs visit virtually all tissues searching for their cognate antigens.29 Furthermore, MBs have been shown to access immune-privileged sites in pathologies like multiple sclerosis and autoimmune orchitis,30,31 consistent with the notion that MBs serve as COO for primary and secondary extranodal DLBCLs.
Cyclic MB reactivation as a pathogenic mechanism
Lymphomagenesis is likely a stepwise process whereby long-lived premalignant clonal precursor cells (CPCs) accumulate genetic and epigenetic lesions over time, ultimately leading to immune-evasive lymphoma phenotypes. Such CPCs may arise from the small subset of MBs that preferentially repopulate new GC reactions after reactivation.26,27 Any genetic or epigenetic perturbation that favors MB commitment to this recall mechanism could foster aberrant outgrowth of MB clones that progressively outcompete NBs and normal MBs at seeding GCs (Figure 2A).24 This skewing would simultaneously force MBs to endure repeated exposure to activation-induced cytidine deaminase (AID)-mediated SHM, concomitantly favoring the accumulation of off-target somatic mutation32 (Figure 2A), a hallmark MCD/C5s.6 Along these lines, Sungalle et al33 showed that sporadic BCL2 overexpression in murine BM cells, mimicking the t(14;18) translocations in follicular lymphoma patients, caused clonal expansion and increased AID-driven mutations in B cells. This phenotype was dependent on repeated antigenic challenge and was attributed to the aberrant survival of BCL2-overexpressing MBs and their cyclic reactivation.33
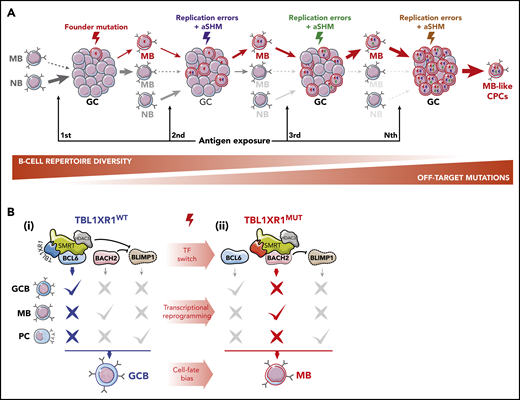
Cyclic MB reactivation as a pathogenic mechanism. (A) GC re-entry as a lymphomagenesis mechanism. Under normal conditions, only a limited subset of MBs partake in new GC reactions after reactivation. In the early stages of malignant transformation, founder mutations, acquired by GCBs as SHM off-target byproducts or resulting from DNA replication errors, can exacerbate this mechanism by producing a set of aberrant MBs that progressively outcompete NB and wild-type (WT) MBs in seeding new GC reactions, favoring their clonal expansion. Concomitantly, participation in successive GC reactions is predicted to result in cumulative acquisition of further off-target mutations in these cells. Such a process is envisioned to take place over long periods of time, ultimately generating an MB-like CPC population. Plasmacytic differentiation, both as another possible GC output and as alternative cell-fate during MB reactivation, is omitted from the scheme for the sake of simplicity but is expected to be impaired by founder or secondary mutations. (B) Epigenetic, transcriptional, and phenotypic reprogramming induced by TBL1XR1 mutations. TFs BCL6, BACH2, and BLIMP1 are required for GCB, MB, and PC development, respectively. (i) In WT GCBs, BCL6 and BACH2 bind to the PRDM1 (BLIMP1) locus and repress its expression, blocking PC differentiation. Transient repression of gene enhancers linked to terminal differentiation by BCL6 and TBL1XR1/SMRT/HDAC3 complexes further prevents MB and PC formation. (ii) TBL1XR1 mutations, as probable founder events, reprogram SMRT/HDAC3 binding from BCL6 to BACH2, causing de-repression of genes required for GCB differentiation, and potentiating the BACH2-driven MB program. Continued repression of PRMD1 by BACH2 maintains GCBs away from the PC fate and shuttles them into an aberrant MB-like state, involved in MCD/C5s early transformation.
Cyclic MB reactivation as a pathogenic mechanism. (A) GC re-entry as a lymphomagenesis mechanism. Under normal conditions, only a limited subset of MBs partake in new GC reactions after reactivation. In the early stages of malignant transformation, founder mutations, acquired by GCBs as SHM off-target byproducts or resulting from DNA replication errors, can exacerbate this mechanism by producing a set of aberrant MBs that progressively outcompete NB and wild-type (WT) MBs in seeding new GC reactions, favoring their clonal expansion. Concomitantly, participation in successive GC reactions is predicted to result in cumulative acquisition of further off-target mutations in these cells. Such a process is envisioned to take place over long periods of time, ultimately generating an MB-like CPC population. Plasmacytic differentiation, both as another possible GC output and as alternative cell-fate during MB reactivation, is omitted from the scheme for the sake of simplicity but is expected to be impaired by founder or secondary mutations. (B) Epigenetic, transcriptional, and phenotypic reprogramming induced by TBL1XR1 mutations. TFs BCL6, BACH2, and BLIMP1 are required for GCB, MB, and PC development, respectively. (i) In WT GCBs, BCL6 and BACH2 bind to the PRDM1 (BLIMP1) locus and repress its expression, blocking PC differentiation. Transient repression of gene enhancers linked to terminal differentiation by BCL6 and TBL1XR1/SMRT/HDAC3 complexes further prevents MB and PC formation. (ii) TBL1XR1 mutations, as probable founder events, reprogram SMRT/HDAC3 binding from BCL6 to BACH2, causing de-repression of genes required for GCB differentiation, and potentiating the BACH2-driven MB program. Continued repression of PRMD1 by BACH2 maintains GCBs away from the PC fate and shuttles them into an aberrant MB-like state, involved in MCD/C5s early transformation.
Follicular lymphomas are indolent tumors reflecting a GCB phenotype.34 Instead, MCD/C5 DLBCLs are highly aggressive tumors composed of malignant MBs that seem to originate at least in part from a distinct and highly aberrant form of MB cyclic re-entry.7 Specifically, focal deletions or somatic mutations in TBL1XR1, a subunit of SMRT/NCOR1 corepressor complexes,35 skew GCB cell fate toward an IgM+ MB population, with increased GC re-entry capacity.7 TBL1XR1 genetic lesions likely represent founder events in MCD/C5 human tumors, as per timing analysis of genetic drivers,6 supporting a role for these alterations in early transformation. TBL1XR1 dysfunction mediates these effects by impairing association of the SMRT/NCOR1 complex with the GCB transcriptional repressor BCL6, instead inducing its binding to the TF BACH2,7 to drive GCBs toward the MB fate36 (Figure 2B). This pro-BACH2 effect simultaneously blocks GCBs from forming PCs by repressing PRDM1,7 a key PC TF37 (Figure 2B).
MCD/C5s and PENLs harbor particularly high AID-driven mutation burden.6,38,39 Accordingly, after repeated antigenic challenges, TBL1XR1-deficient mice develop aggressive MCD/C5-like lymphomas that present very high levels of AID-induced immunoglobulin SHM and off-target mutations in MCD/C5 genes, such as PIM1.7 Notably, extranodal tumors showed the highest burden of mutations compared with nodal tumors in the same animals or Tbl1xr1WT lymphomas.7 These observations support the idea that progressive mutation accumulation in MBs leads to MCD/C5 lymphomagenesis. Importantly, immune profiling of MCD/C5 clinical specimens revealed distinctive MB-like traits, including CD38 downregulation and upregulation of CD27, even in TBL1XR1WT tumors.7 This suggests that different combinations of mutations in these tumors may follow similar transformation paths through expansion and dissemination of malignant MBs.
Antigenic challenges driving MB reactivation and lymphomagenesis
The above mechanism implies that MCD/C5 CPCs require multiple rounds of reactivation. ABC-DLBCLs rely on nuclear factor-κB (NF-κB) signaling for survival and proliferation,40 and MCD/C5s and PENLs present recurrent mutations in Toll-like receptor (TLR) and BCR signaling mediators (eg, MYD88 and CD79B), contributing to the chronic activation of this pathway.5,6 Interestingly, clinical trials with BCR pathway inhibitors also showed BCR signaling dependency in ABC-DLBCLs that do not harbor these mutations,41 suggesting possible involvement of nongenetic mechanisms. In this regard, persistent antigen exposure, in the context of (1) self-reactivity or (2) chronic/recurrent infections, could account for both persistent BCR activation in established tumors and MB reactivation during lymphomagenesis:
BCR activation through a self-reactive component
Autoimmune diseases, such as systemic lupus erythematosus, rheumatoid arthritis, and Sjögren syndrome, are significant risk factors for DLBCL.42,43 Notably, MCD/C5-like mutations were recently identified in B cells producing pathogenic autoantibodies in Sjögren syndrome patients,44 suggesting the involvement of similar CPC populations among these diseases. Further supporting a role for auto-reactivity in MCD/C5 transformation and survival, the BCR repertoires of these tumors are differentially enriched for the self-reactive variant VH4-34.4,45,46 Indeed, BCR activation in MCD/C5 cell lines, critical for survival, was shown to depend on self-reactivity against components of the B-cell membrane, apoptotic debris, or the BCR itself.47 Similarly, increased self-/poly-reactivity has been implicated in primary CNS lymphoma (PCNSL) pathogenesis by facilitating BCR activation by multiple CNS antigens.46
The adaptive immune system normally suppresses self-reactive B cells.48 However, recent studies have found that anergic B cells that recognize both foreign and self-antigens can be activated by immunization and recruited into GCs49 (Figure 3A). These B cells redeem themselves by undergoing SHM and positive selection to reduce their self-reactivity while maintaining the capacity to recognize an exogenous antigen48 (Figure 3A). In the context of MCD/C5 early pathogenesis, aberrant BCL2 overexpression4 may disproportionately spare self-reactive B cells from cell death. Likewise, TBL1XR1 mutations could shunt self-reactive B cells into the MB compartment in the absence of T-cell selection (Figure 3A), because this mechanism occurs even in the absence of follicular T-helper (TFH)-driven CD40L signaling.7
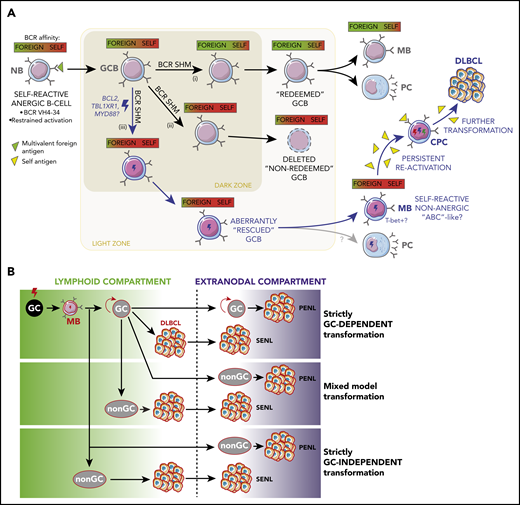
Antigenic drivers and immune context of MB reactivation. (A) Aberrant redemption of self-reactive B cells as an early lymphomagenic event. Anergic B cells whose BCRs recognize both self- and foreign antigens can be recruited into GC reactions if activated by the latter. (i) Once inside the GC, these B cells can redeem themselves and terminally differentiate if BCR SHM causes them to increase their affinity for the foreign antigen while minimizing their reaction against self. (ii) If the edited BCR does not portray a corrected affinity balance or if it acquires a stronger self-reactive character, GCBs are selected to undergo cell death. (iii) However, concomitant somatic mutations outside the BCR locus, such as those targeting BCL2 and TBL1XR1, hold the potential to aberrantly spare self-reactive cells, allowing them to egress the GC, and persist as MBs capable or reactivating, in a fashion reminiscent of aged/autoimmune B cells ("ABCs," discussed in main text). Persistent reactivation of these cells by self-antigens could then sustain the malignant transformation process. (B) Alternative niches and compartments for B-cell malignant transformation. Although founder mutations in primary and secondary extranodal lymphoma (SENLs) most likely occur in the context of canonical GC reactions, further transformation might follow alternative trajectories. Mixed and strictly non–GC-dependent transformation models might better account for the pathogenesis of PENLs, particularly those targeting immune-privileged organs (discussed in main text).
Antigenic drivers and immune context of MB reactivation. (A) Aberrant redemption of self-reactive B cells as an early lymphomagenic event. Anergic B cells whose BCRs recognize both self- and foreign antigens can be recruited into GC reactions if activated by the latter. (i) Once inside the GC, these B cells can redeem themselves and terminally differentiate if BCR SHM causes them to increase their affinity for the foreign antigen while minimizing their reaction against self. (ii) If the edited BCR does not portray a corrected affinity balance or if it acquires a stronger self-reactive character, GCBs are selected to undergo cell death. (iii) However, concomitant somatic mutations outside the BCR locus, such as those targeting BCL2 and TBL1XR1, hold the potential to aberrantly spare self-reactive cells, allowing them to egress the GC, and persist as MBs capable or reactivating, in a fashion reminiscent of aged/autoimmune B cells ("ABCs," discussed in main text). Persistent reactivation of these cells by self-antigens could then sustain the malignant transformation process. (B) Alternative niches and compartments for B-cell malignant transformation. Although founder mutations in primary and secondary extranodal lymphoma (SENLs) most likely occur in the context of canonical GC reactions, further transformation might follow alternative trajectories. Mixed and strictly non–GC-dependent transformation models might better account for the pathogenesis of PENLs, particularly those targeting immune-privileged organs (discussed in main text).
BCR activation through chronic exposure to foreign antigens
Alternatively, long-term BCR stimulation could result from persistent exposure to foreign antigens. For example, chronic hepatitis B (HBV) or C (HCV) virus infections are associated with higher DLBCL incidence.50,51 In the case of HBV, oncogenic effects could result from genetic changes introduced by viral integration into the host genome,52 like in HBV-induced carcinomas.53 However, HCV is incapable of integrating into the host genome, and the BCR repertoires of HBV-positive DLBCLs appear highly restricted to viral antigens and show SHM burden,52 suggesting that tumors arise from the transformation of HBV antigen–selected B cells. Furthermore, the HBV/DLBCL association is not observed in sporadic infections or vaccinations,54 suggesting a requirement for persistent antigen exposure in the transformation process. Despite HV infections being more frequently associated with NOTCH2-mutated DLBCLs,55 whose pathogenesis might differ from MCD/C5s, recent genetic profiling of DLBCLs in patients with concomitant HBV infections56 identified recurrent mutations in PIM1, MYD88, BTG1, and TBL1XR1, suggesting that persistent viral antigen exposure might alternatively favor MCD/C5 pathogenesis.
The self- or foreign antigen scenarios above are not mutually exclusive, because self-reactivity can arise from cross-reactive BCRs originally elicited in responses to pathogens, through molecular mimicry.57 Beyond the nature of the antigens driving BCR activation, a distinctive feature of MCD/C5s and PENLs is their limited class-switch recombination (CSR) and consequent bias toward the IgM isotype.4,45,46,58 This was proposed to lock tumor cells in a state of elevated AID activity while simultaneously preventing complete terminal differentiation.45 Notably, the subset of MBs that avoid PC differentiation and repopulate GCs on reactivation are also largely IgM+,26,27 further suggesting their involvement in MCD-DLBCL pathogenesis.
Are canonical GCs required for MCD/C5 lymphomagenesis?
Another critical question relates to the nature of the immune-niche context where CPCs become activated. Based on their distinctive AID-driven genomic footprint, MCD/C5s are thought to originate from GC-transited B cells.6,46 In this scenario, founder mutations, such as those targeting TBL1XR1, MYD88, or CD79B, would be acquired sporadically by GCBs as byproducts of SHM and proliferation-associated damage.32 Further transformation would require progressive accumulation of these kinds of mutations by MB subsets. Several lines of evidence suggest that this process would require cyclic re-entry into canonical GC reactions (Figures 2A and 3B). First, AID-driven SHM is a GC hallmark, allowing the diversification of BCR repertoires during the adaptive immune response.1 Second, productive synaptic interactions between GCBs and GC-specific stromal populations (ie, TFH and follicular dendritic cells) are required for GCBs to undergo additional rounds of clonal expansion and SHM.59 Third, TBL1XR1 mutant MBs show increased tendency for GC re-entry,7 further supporting GC involvement in pathogenesis.
However, a strictly GC-dependent model might be hard to reconcile with PENLs, which develop without apparent prior growth in lymphoid organs, suggesting that at least ulterior transformation steps occur at extranodal, and sometimes immune-privileged, sites (Figure 3B). Recent studies have identified circulating TFH populations that originate in lymph nodes and disseminate60-62 and could potentially fulfill functions similar to their follicular counterparts at extranodal sites.63 Although T-helper cells and antigen-presenting cells are present in some PCNSLs,64 their characteristics and functions have not been elucidated. Moreover, despite the observation of follicle-like structures in the leptomeninges of patients with advanced multiple sclerosis,65 definite evidence of ectopic GCs at immune-privileged sites is lacking. As further counterpoint to a GC-driven model, TBL1XR1 mutations actually impair the GC reaction by disrupting BCL6 function,7 hinting that acquisition of a mature GCB profile is dispensable for transformation.
If MCD/C5 and PENL transformation does not require B cells partaking in canonical GCs, in which context are these cells exposed to AID activity? Interestingly, it was recently shown that CSR, an AID-catalyzed mechanism, occurs primarily before activated B cells form GCs.66 Additionally, AID can be detected in highly proliferating extrafollicular B cells.67 Furthermore, hypermutated non–GC-transited MBs have been identified in patients harboring germline mutations that block CD40L expression in T cells68 (required for B-cell entry into GCs) and in extrafollicular regions of lymphoid tissues in autoimmunity models.69 These studies indicate that antigen and T-cell–like driven signals, but not follicular dendritic cells and TFH themselves, are required to support AID function. Along these lines, alternative populations provide stromal support during immune responses, such as neutrophil B-cell helper cells that facilitate marginal zone B-cell activation.70 Given that immune-privileged sites restrict lymphocytic access, one could envision a scenario where local stromal populations, acting as unorthodox surrogates, could support MB-cell reactivation, extranodal SHM, and transformation. Indeed, CD40L blockage in mice extinguishes GCs71 but disproportionately spares Tbl1xr1-mutant MB formation,7 suggesting that TFH might be dispensable for transformation. Further supporting this observation, transcriptomic analysis of MCD/C5 tumors showed depletion for CD4+ and TFH signatures.4
It should be noted that these GC and non-GC activation models are again not necessarily mutually exclusive, because GC re-entry may be required to establish early MB-like CPC populations prone to reactivate but dispensable for further mutation accumulation and overt transformation (Figure 3B). The next section will address what those later transformation steps might look like and how these would account for MCD/C5s phenotypic presentation.
A putative oscillatory mechanism may generate the complex MCD/C5 phenotype
MCD/C5 development involves MB cyclic reactivation and AID-driven mutagenesis but may not necessarily require re-entry into canonical GCs. It is then logical to consider whether and how these processes could be accelerated to foster repeated rounds of activation and mutagenesis. In normal B cells, strong antigen and mitogen stimulation activate AID transcription,72 in a process partially mediated by the TF IRF4.15,73 Activating mutations in CD79B, PIM1, or MYD88 might confer lower activation thresholds to BCR and TLR engagement in CPCs that could trigger NF-κB signaling40 and IRF4 upregulation, ultimately leading to bursts of proliferation and AID activity.74 Indeed, MCD/C5s express elevated levels of IRF4 and its targets,75 and ABC-DLBCs exhibit high AID expression.74 IRF4 also normally induces PC differentiation15,73 by repressing BCL6 and inducing PRDM1 (Figure 4A). This latter aspect of IRF4 function, and CPC differentiation beyond an early PB state, might be blocked in MCD/C5s by inactivating mutations targeting PRDM1,4 or through persistent BACH2-driven PRDM1 transcriptional repression after TBL1XR1 mutations7 (Figure 4B).
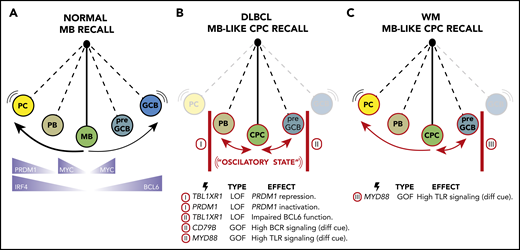
An oscillatory lymphomagenesis circuit model. (A) On reactivation, normal MBs undergo terminal plasmacytic differentiation or, less frequently, populate new GC reactions. Key TFs involved in these processes are highlighted at the bottom of the scheme. (B) During MCD/C5 and PENLs transformation, MB-like CPCs are proposed to get caught in an oscillatory lymphomagenic state. In this scenario, CPCs with low TLR and BCR signaling thresholds become repeatedly reactivated, but unlike normal MBs, their complete differentiation into PC or GCB is blocked by somatic mutations (as exemplified at the bottom of the scheme). Hence, CPCs instead reversibly swing between early PB and pre-GCB states, undergoing bursts of proliferation and AID activation, favoring the acquisition of ulterior mutations needed to achieve immune evasion, and accounting for the aberrant MB-like presentation of these tumors. (C) WM malignant transformation may follow similar or identical initial trajectories than MC/C5s by forming MB-like CPCs. However, somatic mutations in WM are expected to block the GCB cell fate but allow plasmacytic differentiation, accounting for the distinctive LLPC-like presentation of these tumors.
An oscillatory lymphomagenesis circuit model. (A) On reactivation, normal MBs undergo terminal plasmacytic differentiation or, less frequently, populate new GC reactions. Key TFs involved in these processes are highlighted at the bottom of the scheme. (B) During MCD/C5 and PENLs transformation, MB-like CPCs are proposed to get caught in an oscillatory lymphomagenic state. In this scenario, CPCs with low TLR and BCR signaling thresholds become repeatedly reactivated, but unlike normal MBs, their complete differentiation into PC or GCB is blocked by somatic mutations (as exemplified at the bottom of the scheme). Hence, CPCs instead reversibly swing between early PB and pre-GCB states, undergoing bursts of proliferation and AID activation, favoring the acquisition of ulterior mutations needed to achieve immune evasion, and accounting for the aberrant MB-like presentation of these tumors. (C) WM malignant transformation may follow similar or identical initial trajectories than MC/C5s by forming MB-like CPCs. However, somatic mutations in WM are expected to block the GCB cell fate but allow plasmacytic differentiation, accounting for the distinctive LLPC-like presentation of these tumors.
Early precursor (pre)-GCBs express MYC and AID,66,76 as well as low-intermediate levels of BCL677 (Figure 4A). MYC is required for B cells to accumulate metabolic precursors and biomass to support their proliferative bursting, but its expression is repressed in GCBs by their higher BCL6 levels.76 MCD/C5s feature upregulation of MYC target genes and proliferation programs,4 as well as AID and BCL6, consistent with their putative origin from activated MB cells that can alternatively access a pre–GCB-like status. The fact that many GCB BCL6 target genes are derepressed in MCD/C5s,4 perhaps because of mutation-driven immune synapse-like signaling in CPCs or to TBL1XR1 somatic mutations disrupting BCL6 function,7 further suggests that acquisition of a mature GCB profile is blocked during transformation. Indeed, despite harnessing proto-oncogenic features from both early PB and pre-GCB populations, transcriptional profiles of MCD/C5s are depleted for both mature GCB and PC signatures.4
A plausible interpretation of these observations is an oscillatory lymphomagenesis circuit (Figure 4B) whereby highly labile MB-like CPCs, with reduced activation threshold caused by TLR and BCR activating mutations, undergo repeated rounds of activation-associated induction of MYC, AID, and perhaps BCL6. However, full engagement of the BCL6 program is impaired in these cells by mutations such as those in TBL1XR1 that drive CPCs back into the MB cell state while also preventing terminal PC differentiation (Figure 4B). The resulting tumors would then reflect the final stages of this process: that of transformed MBs trapped in an activated state.
The oscillatory model proposed here could similarly account for the transformation and presentation of other MB-derived lymphomas, like Waldenström macroglobulinemia (WM) (Figure 4C). WMs are mostly indolent but share features with MCD/C5s. (1) More than 90% of WMs harbor MYD88L265P, the same variant found in MCD/C5s.78 (2) WM pathogenesis has been linked to autoimmune disorders and chronic infections.79 (3) WMs exhibit SHM burden and express CD27.79,80 (4) WMs show limited CSR.79 (5) Approximately 10% of WMs can transform into DLBCLs, 80% of which show extranodal involvement.81 However, most WMs are characterized by the accumulation of lymphoplasmacytic cells in the BM and exacerbated monoclonal IgM secretion79 in a fashion more reminiscent of LLPCs than MBs. These observations raise the possibility that WMs could initially follow a common trajectory with MCD/C5s but that the absence of mutations preventing them from oscillating toward plasmablastic phenotypes (like those targeting TBL1XR1 or PRDM1; Figure 4C) would result in ulterior differences in tumor presentation.
An aged/autoimmune B-cell origin for ABC-DLBCLs?
Defining CPC phenotypic traits would constitute a significant step toward the early detection of MCD/C5s and PENLs, considering these could potentially even be screened for in circulation. MBs prone to repopulate GCs are mostly encompassed within CD27+IgM+IgD− B cells,26,27 a minor population in peripheral blood and spleen.82 However, even these relatively rare cells are diverse and heterogeneous in terms of their origin, antigen experiences, and function. Intriguingly, re-entry–prone MBs prospectively giving rise to MCD/C5s7 bear notable similarity to aged/autoimmune B cells.83,84 Aged/autoimmune B cells are observed in elderly female mice and in humans and mice infected with specific pathogens or with autoimmune disorders.85 Beyond differences in their phenotypic definition,85 aged/autoimmune B cells stain positive for CD11c and express and depend on the TF T-BET (TBX21).86 Much like their normal MB counterparts, aged/autoimmune B cells can follow various cell fates on activation, such as entering GCs, forming PCs or MBs, or self-renewing.87
Additional factors link aged/autoimmune B cells to MCD/C5s. (1) SHM burden identifies aged/autoimmune B cells as GC-transited cells.88 (2) Aged/autoimmune B cells accumulate in the context of persistent BCR activation by chronic infections or self-reactivity.83,89 (3) T-BET induction and aged/autoimmune B cells development and survival are critically dependent on MYD88 signaling.83,90 (5) Despite T-BET’s role in promoting CSR, a significant fraction of aged/autoimmune B cells remain IgM+.85 (4) Aged/autoimmune B cells produce and secrete interleukin-10,84 similar to ABC-DLBCLs.91 These considerations raise the possibility that MCD/C5 DLBCLs may develop from MB-like CPCs that are similar to, or indistinguishable from, aged/autoimmune B cells, as further suggested by the presence of canonical ABC-DLBCL somatic mutations in MBs of patients with autoimmune disorders.44 These findings highlight the need to identify features that might be predictive of aged/autoimmune B cells/MB/CPCs transformation to overt MCD/C5s and PENLs.
Clonal expansion under the (immune) radar
Avoiding immune eradication by becoming undetectable or resistant to cytotoxic attacks is a prerequisite for lymphomagenesis. However, normal MBs are highly attuned to the immune system, scouting tissues for foreign antigens and closely collaborating with T cells.29 Such behavior would presumably restrict the ability of MB-like CPCs to grow in an unrestricted manner, meaning they must find ways to elude T-cell surveillance and clearance. On the one hand, lymphomas targeting immune-privileged sites might circumvent controls by taking advantage of the selective access granted by these organs to immune cells.92 In other words, malignant precursors that manage to infiltrate these sites, as MBs are known to do in autoimmune disorders,30,31 could harness natural barriers to partially isolate themselves from immune surveillance. However, this is not enough to completely avoid immune clearance,64 and most MCD/C5s exist outside of immune-privileged sites,4,6 indicating that additional strategies must come into play. Along these lines, it was shown that ∼75% of MCD/C5s acquire genomic lesions that reduce antigen presentation (through MHC-I or TAP1 inactivation) or directly impairing natural killer and T-cell activation (through CD58 inactivation and PD-L1 or PD-L2 gene fusions and overexpression).4 The high prevalence of these alterations suggests these are critically needed for CPCs to give rise to full-blown lymphomas. Beyond these direct hits, MYD88 activating mutations can also contribute to immune evasion by inducing the expression of the T-cell inhibitory molecule PD-L1.93 Additionally, MYD88 genomic lesions could enhance production of interleukin-10,94 whose expression is elevated in MCD/C5s.4 This cytokine promotes autocrine tumor growth through JAK2/STAT3 activation91 but can also restrict proinflammatory cytokine production, costimulatory molecule expression, and antigen presentation,95 ultimately terminating T-cell responses and facilitating tumor immune evasion.
Implications of an MB COO for targeted therapies
Although R-CHOP (rituximab, cyclophosphamide, doxorubicin hydrochloride [hydroxydaunomycin], vincristine sulfate [oncovin], and prednisone) significantly improved overall DLBCL prognosis, 30% to 50% of patients show resistance or relapse after treatment,96 with MCD/C5s showing the worst response and outcome.5,6 The clinical management of extranodal DLBCLs presents additional challenges, given their particular anatomic localization,97 highlighting the need for rationally designed subtype-specific treatments. BCR-dependent NF-κB activation in MCD/C5s has prompted interest in agents targeting this pathway, such as ibrutinib and lenalidomide.98 Ibrutinib targets BTK kinase, which links BCR and TLR/MYD88-driven NF-κB activation, and clinical trials with this drug have shown promising results for MCD/C5s41 and PCNSL.99,100 However, BTK inhibitors alone do not control MCD/C5s, making it necessary to target additional biological vulnerabilities in these tumors. One approach to circumventing signaling bypass mechanisms is to target additional nodes of the BCR and MYD88 signaling pathways, such as IRAK4,101 MALT1,102,103 and PI3K/mTOR.100,104
Complementary to these strategies, an MB COO for extranodal tumors provides alternative targets. The histone deacetylase HDAC3 collaborates with BCL6 in shaping the chromatin landscape and transcriptome of GCBs and GCB-DLBCLs.105-107 Accordingly, selective inhibition of HDAC3 was shown to be an efficient targeted therapy against GCB-DLBCLs harboring mutations in the histone acetyltransferase CREBBP.108 TBL1XR1 mutation drives MCD/C5s through aberrant recruitment of SMRT/HDAC3 complexes to BACH2,7 suggesting that MCD/C5s might also be dependent on HDAC3 function and sensitive to its inhibition. Interleukin-9R (IL-9R) is another potentially interesting target, because it was shown to mediate the aberrant expansion of MB precursors after TBL1XR1 genetic lesions.7 Additional studies have implicated IL-9/IL-9R signaling in MB recall responses109 and found a correlation between IL-9R overexpression and worse clinical outcome in a small DLBCL cohort.110 Last, chemokine receptors mediating the homing of CPCs and tumor cells to extranodal sites remain largely unexplored but hold great potential for approaches aimed at blocking dissemination.
As mentioned previously, cytotoxic therapy resistance and relapse are prevalent among extranodal lymphomas. Although particular mechanisms may vary, these phenomena could stem from the MB-like character of these tumors. First, the epigenetic and transcriptional plasticity of MBs could be harnessed by tumor cells to acquire molecular resistance mechanisms to therapies, even in the absence of additional somatic mutations. Second, a fraction of CPCs could remain or become dormant/quiescent, like resting MBs, generating a reservoir that withstands the ablation of highly proliferative cells, and is thus able to repopulate the tumor and contribute to the relapsing nature of these disease in a fashion similar to stem cell populations in leukemias. Future research endeavors should then focus on identifying these CPCs, understanding their niche, and elucidating ways to eradicate them.
Concluding remarks
Synthesizing data discussed above, we propose a modified stepwise model for MCD/C5s and PENLs pathogenesis through the progressive malignant transformation of MB cells (Figure 5). A deeper exploration of MB subpopulations and their aberrant dysregulation is likely to improve our understanding of these diseases and holds additional potential for elucidating the partially overlapping nature of DLBCLs and other immunologic disorders. Collectively, these observations invite us to rethink the way we conceive, diagnose, and treat these often fatal diseases.
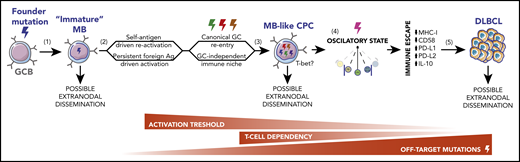
Stepwise model for MCD/C5s and PENLs pathogenesis. (1) During normal immune responses, SHM or DNA replication errors stochastically introduce off-target founder mutations in GCBs, favoring the development of an immature long-lived MB population with increased tendency to acquire a GCB-like profile on recall, at the expense of terminal PC differentiation. (2) Cyclic reactivation of these MBs over extended periods of time favors their clonal expansion and increases the chances of acquiring additional off-target mutations. Repeated activation may be driven by a self-reactive BCR or chronic exposure to foreign agents and could occur in the context of canonical GCs or in a GC-independent manner. (3) Founder or secondary-acquired mutations, targeting BCR and TLR pathway mediators, lower the immune activation threshold of these MB-like CPCs, driving them into a semipersistent activated state. At this stage, cells are predicted to become less dependent on canonical costimulatory signaling. (4) Persistent activation in the context of MBs intrinsic phenotypic plasticity traps CPCs in a nearly cell-autonomous oscillatory state, navigating between MB-like, pre-GC–like, and PB-like phases, whereas somatic mutations block full lineage commitment. (5) These CPCs intermittently undergo bursts of proliferation and AID activation, allowing the acquisition of ulterior genomic lesions that, paired with transcriptional/epigenetic remodeling, enable complete immune evasion and overt tumor development.
Stepwise model for MCD/C5s and PENLs pathogenesis. (1) During normal immune responses, SHM or DNA replication errors stochastically introduce off-target founder mutations in GCBs, favoring the development of an immature long-lived MB population with increased tendency to acquire a GCB-like profile on recall, at the expense of terminal PC differentiation. (2) Cyclic reactivation of these MBs over extended periods of time favors their clonal expansion and increases the chances of acquiring additional off-target mutations. Repeated activation may be driven by a self-reactive BCR or chronic exposure to foreign agents and could occur in the context of canonical GCs or in a GC-independent manner. (3) Founder or secondary-acquired mutations, targeting BCR and TLR pathway mediators, lower the immune activation threshold of these MB-like CPCs, driving them into a semipersistent activated state. At this stage, cells are predicted to become less dependent on canonical costimulatory signaling. (4) Persistent activation in the context of MBs intrinsic phenotypic plasticity traps CPCs in a nearly cell-autonomous oscillatory state, navigating between MB-like, pre-GC–like, and PB-like phases, whereas somatic mutations block full lineage commitment. (5) These CPCs intermittently undergo bursts of proliferation and AID activation, allowing the acquisition of ulterior genomic lesions that, paired with transcriptional/epigenetic remodeling, enable complete immune evasion and overt tumor development.
Acknowledgments
This work was supported by grants from the Leukemia & Lymphoma Society (Career Development Program Grant 5469-18 to L.V., LLS-TRP-6572-19 and LLS-SCOR-7012-16 to A.M.M.), the National Cancer Institute (NCI-R35-CA220499 to A.M.M.), the Chemotherapy Foundation (to A.M.M.), and the Follicular Lymphoma Consortium (to A.M.M.). Cartoons used in figures were adapted from images in the Servier Medical Art repository, within the terms of the Creative Commons Attribution 3.0 Unported License.
Authorship
Contribution: L.V. and A.M.M. conceptualized and prepared the figures and manuscript.
Conflict-of-interest disclosure: A.M.M. receives research funding for Janssen and Sanofi, is on the scientific board of KDAC pharmaceuticals, and has consulted for Constellation, Epizyme and Jubilant. L.V. declares no competing financial interests.
Correspondence: Ari M. Melnick, Division of Hematology/Oncology, Department of Medicine, Weill Cornell Medicine, 413E 69th St, New York, NY 10021; e-mail: amm2014@med.cornell.edu; and Leandro Venturutti, Division of Hematology/Oncology, Department of Medicine, Weill Cornell Medicine, 413E 69th St, New York, NY 10021; e-mail: lev2009@med.cornell.edu.
