

In this issue of Blood, show that multiple myeloma CD38 expression is inhibited by the bone marrow microenvironment, in particular interleukin-6 (IL-6)/signal transducer and activator of transcription 3 (STAT3) signaling, resulting in less effective targeting by monoclonal antibodies.1
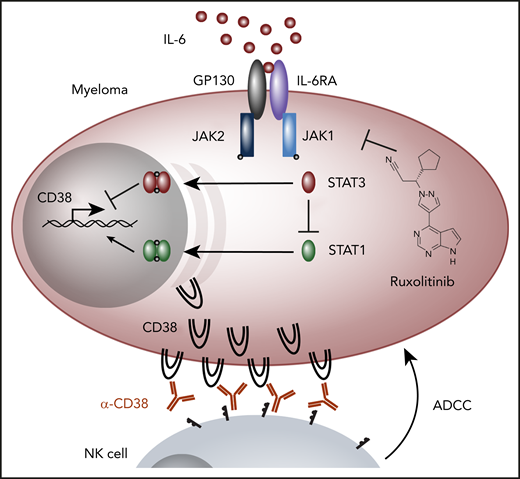
IL-6 signaling in myeloma and CD38 regulation. The cytokine IL-6 binds the receptors IL-6 receptor alpha (IL-6RA) and GP130 to activate JAK1 and JAK2 kinases that phosphorylate STAT3 and to a lesser extent STAT1, resulting in their dimerization and nuclear translocation. Ogiya et al find that STAT3 inhibits CD38 expression, whereas STAT1 increases it, and STAT3 knockout results in an upregulation and activation of STAT1. Increased CD38 expression results in more ADCC. Lines with arrows or blocks do not necessarily represent direct interactions. NK, natural killer.
IL-6 signaling in myeloma and CD38 regulation. The cytokine IL-6 binds the receptors IL-6 receptor alpha (IL-6RA) and GP130 to activate JAK1 and JAK2 kinases that phosphorylate STAT3 and to a lesser extent STAT1, resulting in their dimerization and nuclear translocation. Ogiya et al find that STAT3 inhibits CD38 expression, whereas STAT1 increases it, and STAT3 knockout results in an upregulation and activation of STAT1. Increased CD38 expression results in more ADCC. Lines with arrows or blocks do not necessarily represent direct interactions. NK, natural killer.
Monoclonal antibodies have changed the treatment landscape in multiple myeloma by adding a third “backbone” to the standard treatment approaches, resulting in deeper and more durable responses than those achieved with steroids, alkylators, proteasome inhibitors, or immunomodulatory agents. Daratumumab and isatuximab target CD38, an adenosine 5′-diphosphate-ribose hydrolase that regulates calcium signaling. CD38 is expressed at high levels on myeloma cells but is also expressed on T cells, B cells, and natural killer cells. While targeting CD38 clearly improves patient outcomes, monotherapy in refractory disease has limited efficacy.2 It has also been demonstrated that CD38 expression is linked to the degree of antibody-dependent cellular cytotoxicity (ADCC).3 Here, Ogiya et al show how the microenvironment can regulate CD38 expression which potentially impacts the therapeutic efficacy of this target.1 Using a focused panel of cytokines, they identify the culprit as IL-6, a cytokine that promotes plasma cell differentiation, myeloma cell growth, and drug resistance.4 They also found that interferon-β (IFN-β) and IFN-γ increase CD38 expression, consistent with previous reports.5
To pinpoint the mechanism by which IL-6 downregulates CD38 expression, they performed a genome-wide clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 screen under IL-6 stimulation comparing the top 5% of CD38-expressing cells to others. This identified STAT3 and Janus kinase 1 (JAK1), well-established signaling mechanisms downstream of the IL-6 receptor (see figure), as negative regulators of CD38 expression. STAT3 was investigated using both CRISPR-Cas9 knockout and short interfering RNA knockdown isogenic cell lines revealing a complete abrogation of bone marrow supernatant-mediated downregulation of CD38. Furthermore, expression of a constitutively active STAT3 also downregulated CD38. In the absence of STAT3, IL-6 substantially upregulated and activated STAT1, resulting in a subtle increase in CD38 (see figure). To therapeutically combat IL-6–mediated CD38 downregulation, the authors employed the JAK inhibitor ruxolitinib that targets both JAK1 and JAK2, thereby disrupting the signaling cascade from the IL-6 receptor (see figure). Ruxolitinib inhibited both STAT3 and STAT1 activation, causing upregulation of CD38, and enhanced daratumumab-induced ADCC against myeloma cells in the presence of bone marrow supernatant. This trend was also seen in patient samples, although the responses were more heterogeneous.
Ogiya et al clearly demonstrate that the bone marrow supernatant–derived IL-6/STAT3 signaling pathway downregulates CD38 at transcriptional and protein levels, but the precise mechanism on how this occurs is still elusive. STAT proteins are generally regarded as activators of transcription, suggesting an indirect effect. However, chromatin immunoprecipitation–sequencing data from the ENCODE project show both STAT1 and STAT3 binding the CD38 promoter in lymphoblastoid cells. Elucidating the precise molecular mechanism of STAT3-mediated CD38 downregulation may provide alternative therapeutic strategies for enhancing CD38 expression and daratumumab efficacy.
Perhaps clues into the mechanism of STAT3-mediated CD38 downregulation may be provided by further interrogation of results from the genome-wide CRISPR-Cas9 screen. Interestingly, 7 of the top 10 guide RNAs that negatively regulated CD38 expression target genes on chromosome 19. Approximately half of myeloma cases have trisomy of chromosome 19 as well as most other odd-numbered chromosomes, a phenomenon called hyperdiploidy. The other top hits target genes on chromosome 17 (STAT3), also a trisomy found in hyperdiploid myeloma, and chromosome 1p (JAK1 and ARID1A), which is deleted in smaller fraction of myeloma cases. Thus, one might hypothesize that hyperdiploid myeloma expresses less CD38 while del(1p) myeloma expresses more CD38. These results, combined with characterization of CD38 expression across myeloma subtypes, may help inform the patient populations where CD38 monoclonal antibodies are most effective.
IL-6 clearly results in downregulation of CD38, but it will be important to place these results in the context of other backbone therapies. For instance, at our institution, patients now receive front-line daratumumab added to the very effective combination of bortezomib, lenalidomide, and dexamethasone.6 Immunomodulatory drugs (IMiDs) such as lenalidomide have been reported to upregulate CD38 through an IFN response.7 Similarly, all-trans retinoic acid (ATRA), histone deacetylase (HDAC) inhibitors, and DNA methylation inhibitors have been shown to upregulate CD38, although these agents are less commonly used in myeloma.3,8,9 It is important to know if these therapeutics, especially IMiDs, can overcome the IL-6/STAT3-mediated downregulation of CD38. In this regard, ruxolitinib may not be the best combination for maximizing CD38 expression, because JAK1 and JAK2 are necessary transducers for IFN signaling, which induces CD38 expression. Alternatively, one could more specifically inhibit IL-6 signaling using a monoclonal antibody blockade of IL-6 (siltuximab) or the IL-6 receptor (tocilizumab).
In the context of published data suggesting HDAC inhibitors and ATRA can upregulate CD38 expression and enhance ADCC in vitro, how is this different? Furthermore, how, given that there is no conclusive evidence that upregulation using either of those approaches translates into clinically meaningful responses, is the approach of targeting JAK/STAT different? Perhaps the difference lies in the interplay between the microenvironment and the tumor cells as opposed to wholesale upregulation of CD38 on all cells. A concern using agents such as ATRA is that they upregulate CD38 expression on all cells, potentially increasing off-target toxicity. Here, the authors show the mechanism at play is localized to the tumor environment impacting CD38 expression. Concentrating on IL-6 may not only enhance CD38 targeted therapy but also sensitize to other agents being used in combination.10 Fleshing out the effects of this backbone and other commonly used drugs in context with the complex cellularity of the bone marrow microenvironment is a daunting task but important for rationally designing the best treatment regimens with respect to CD38 monoclonal antibodies. Ogiya and colleagues have identified an important factor regulating CD38 expression, worthy of consideration in future studies, and potentially opening the door to improved CD38 monoclonal antibody efficacy.
Conflict-of-interest disclosure: S.L. is a consultant for Takeda, Celgene, Amgen, Novartis, BMS, Sanofi, and Janssen, as well as director for TG therapeutics. B.G.B. declares no competing financial interests.
