

In this issue of Blood, have demonstrated that acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike proteins activate complement by engaging the alternative pathway (AP) and that blocking this process by inhibiting factor D or C5 could mitigate SARS-CoV-2–induced immunopathology.1
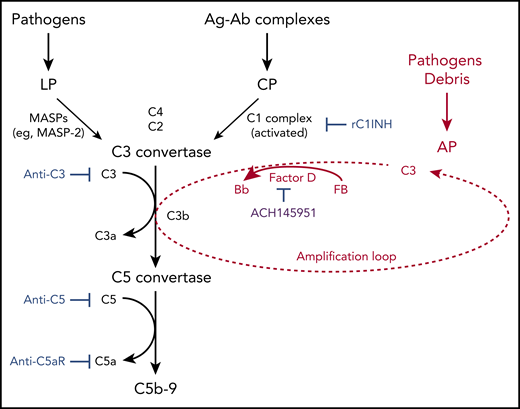
Schematic diagram summarizing clinical trials targeting different aspects of the complement system, registered on www.clinicaltrials.gov as of 3 September 2020. C3 with a cleaved thioester bond (ie, C3b or C3(H2O)) binds factor B. Factor B is then cleaved by factor D to form C3bBb. Properdin (not shown) stabilizes this C3 convertase, which can cleave many molecules of C3 to C3b and thereby amplifies the AP. The factor D inhibitor tested in the paper by Yuan et al (ACH145951) is in purple and is not currently registered. Ag-Ab, antigen-antibody; C5aR, C5a receptor; CP, classical pathway; LP, lectin pathway; MASP, mannan binding lectin-associated serine proteases; rC1INH, recombinant C1-esterase inhibitor. Created with www.biorender.com. For additional information, see Figure 6 in the article by Yu et al that begins on page 2080.
Schematic diagram summarizing clinical trials targeting different aspects of the complement system, registered on www.clinicaltrials.gov as of 3 September 2020. C3 with a cleaved thioester bond (ie, C3b or C3(H2O)) binds factor B. Factor B is then cleaved by factor D to form C3bBb. Properdin (not shown) stabilizes this C3 convertase, which can cleave many molecules of C3 to C3b and thereby amplifies the AP. The factor D inhibitor tested in the paper by Yuan et al (ACH145951) is in purple and is not currently registered. Ag-Ab, antigen-antibody; C5aR, C5a receptor; CP, classical pathway; LP, lectin pathway; MASP, mannan binding lectin-associated serine proteases; rC1INH, recombinant C1-esterase inhibitor. Created with www.biorender.com. For additional information, see Figure 6 in the article by Yu et al that begins on page 2080.
Complement activation occurs in critically ill patients with SARS-CoV-2 infection and likely contributes to end-organ failure.2 A role for the complement system in the pathogenesis of SARS-CoV-2 infection (ie, COVID-19) was suspected based on the similarities to the clinical presentation and the autopsy findings observed in thrombotic microangiopathies: paroxysmal nocturnal hemoglobinuria, atypical hemolytic uremic syndrome, and catastrophic antiphospholipid syndrome, in which the AP plays a key role.2,3 These observations, plus several model systems pointing to a role for the lectin pathway, resulted in the off-label use of complement inhibitors in COVID-19.2 Currently, both phase 2 and 3 studies targeting components of the complement cascade to treat COVID-19 have been initiated (see figure).2 However, many gaps remain in our understanding of why and how SARS-CoV-2 induces an apparent endotheliopathy, including the mechanism(s) by which the virus engages complement.
In addition, multiple studies have reported increased complement activation products in the plasma and/or their deposition in target organs of patients with COVID-19, but which of the 3 pathways of complement activation is primarily responsible, the classical, lectin, or the alternative (see figure), remains unanswered.2,4
Yu et al used an in vitro system employing recombinantly synthesized COVID-19 proteins to ask if the spike proteins in SARS-CoV-2 (ie, S1 and S2) or the N protein–induced complement mediated cytotoxicity. The surprising and major finding in this report is that the purified spike proteins, produced in bacterial or insect cells, activate the AP in buffers containing human serum and a cell line lacking glycolipid anchored proteins. The authors employed this membrane perturbing cell model system along with flow cytometry to demonstrate that SARS-CoV-2 spike proteins bound heparan sulfate on the cell surface and then triggered the AP (see Figure 6 in the Yu et al paper).
Using a small molecule pharmacologic inhibitor of factor D (a plasma serine protease that cleaves factor B to Ba and Bb to form the AP convertase; see figure) and a monoclonal antibody that blocks C5 (cleavage of which results in C5a, a potent anaphylatoxin, and C5b, which initiates formation of the membrane attack complex, C5b-9), the authors inhibited complement activation. Of interest, factor D inhibition was more impressive compared with that for the anti-C5 mAb (based on the fact that the factor D blocker worked earlier in the cascade; ie, inhibited C3 as well as C5 by shutting down the AP). These in vitro results led the investigators to propose that SARS-CoV-2 induces complement activation primarily through the AP and that targeting components of the cascade upstream of C5 (eg, factor D inhibition) may be a more appealing approach to reducing complement-mediated tissue injury in COVID-19 (a concept also suggested by others).5
These studies by Yu et al have a high therapeutic potential and are a welcome advance to the field. However, there are caveats to be considered while building upon this work. First, factor D is one of several proteases that can facilitate complement activation by the AP in vivo. Kallikrein, another serine protease, also cleaves factor B.6 Moreover, we have previously shown that a very low amount of factor D is capable of activating the AP.7 Hence, a better choice for the target in the AP for inhibition may be C3, factor B, or properdin (which augments the AP’s feedback loop). Second, their findings do not explain the interaction between the N protein of coronaviruses and membrane-associated serine proteases that activate the lectin pathway.8 Also, a prior study has suggested that C3-deficient mice were better protected from the consequences of SARS-CoV MA15 infection compared with C4 or factor B-deficient mice, implicating multiple arms of the complement cascade in SARS-CoV–mediated disease.9 Third, although inhibiting complement activation proximal to C5 is inherently attractive, many of the proteins in the proximal aspect of the cascade, such as C3 and factor B, are a part of the acute phase response and may play an important role in the initial control of the infection.2,4,9 Fourth and probably most critical, the study was primarily conducted entirely using an in vitro model system, and, although it points out a potential novel mechanism of SARS-CoV-2–mediated complement activation, it will need to be rigorously examined in in vivo model systems of SARS-CoV-2–mediated immunopathogenesis. In addition, the cells used in their assays (ie, TF1PIGAnull cells) lacked several membrane complement regulatory proteins, and therefore, may not be representative of cell populations in vivo. Likewise, the overall structure of the soluble spike proteins may not be identical to that bound to the virus.
The experiments by Yu et al also demonstrate that the addition of factor H (a plasma regulator of complement activation) neutralizes the effects of the SARS-CoV-2 spike proteins by blocking AP activation in their model system. This result fits with the current hypothesis that some patients with severe COVID-19 may have a defect in complement regulatory proteins.10 Although there are no reports currently that patients with severe COVID-19 infection have lower levels of factor H, a recent article focusing on this patient population identified genetic and/or transcriptional variations in complement (and coagulation) components, including factor H.10 Hence, whether there is a deficiency of factor H in these patients, or a “resistance,” wherein the spike protein blocks factor H from binding to certain glycosaminoglycans, such as heparan sulfate, or affects oligosaccharide conformation, and thus, its protective effects, needs to be explored.
As we develop an improved understanding of the pathogenesis of SARS-CoV-2, including identifying predisposing genetic and environmental risk factors, dissecting specifically how complement activation contributes to its pathogenesis (as Yu et al have done) has obvious implications for designing improved therapeutics. The goal is to halt the deleterious consequences of an uncontrolled immune response without compromising host defense, thereby reducing the morbidity and mortality of this devastating illness.
Conflict-of-interest disclosure: H.S.K. and J.P.A. are coinvestigators on a phase 3 study of ravulizumab in COVID-19 sponsored by Alexion Pharmaceuticals. J.P.A. reports serving as a current consultant for Celldex Therapeutics, Clinical Pharmacy Services, Kypha Inc, Achillion Pharmaceuticals, and BioMarin Pharmaceutical and receives stock or equity options in Compliment Corporation, Kypha Inc, Gemini Therapeutics Inc, and AdMiRx Inc.
