

After decades of dissecting the signaling pathways that trigger and amplify platelet activation, we still know very little about the regulatory mechanisms that limit and control these processes. In this issue of Blood, shed light on the importance of RGS10 and RGS18, 2 molecular brakes of the regulator of G protein signaling (RGS) family, in modulating platelet activation and platelet number.1
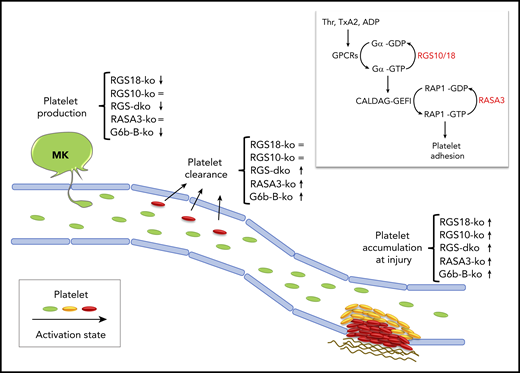
In vivo phenotype of mice lacking RGS10/18 or other platelet molecular brakes. Key features of the in vivo phenotype of mice lacking critical molecular brakes of platelets are (1) increased platelet accumulation at sites of injury, (2) increased platelet clearance, and (3) thrombocytopenia. Double deficiency of RGS10 and RGS18 results in an exaggerated thrombus growth and in a mild thrombocytopenia, which is in part due to increased platelet clearance, and in part due to a nonredundant contribution of RGS18 to the regulation of platelet production. The phenotype of other molecular brakes, RASA3 and G6b-B, is shown for comparison. Symbols indicate whether a knockout mouse (ko) displays an increased (↑), decreased (↓), or unaltered (=) phenotype. The inlet shows the 2 main G protein switches that control rapid agonist-induced platelet adhesion: (1) RGS10/18 negatively regulates the heterotrimetic G proteins coupled to surface receptors (GPCRs) stimulated by soluble agonists such as thrombin (Thr), thromboxane A2 (TxA2), and adenosine diphosphate (ADP); (2) RASA3 negatively regulates the monomeric G protein RAP1, critical regulator of integrin-mediated adhesion. MK, megakaryocyte.
In vivo phenotype of mice lacking RGS10/18 or other platelet molecular brakes. Key features of the in vivo phenotype of mice lacking critical molecular brakes of platelets are (1) increased platelet accumulation at sites of injury, (2) increased platelet clearance, and (3) thrombocytopenia. Double deficiency of RGS10 and RGS18 results in an exaggerated thrombus growth and in a mild thrombocytopenia, which is in part due to increased platelet clearance, and in part due to a nonredundant contribution of RGS18 to the regulation of platelet production. The phenotype of other molecular brakes, RASA3 and G6b-B, is shown for comparison. Symbols indicate whether a knockout mouse (ko) displays an increased (↑), decreased (↓), or unaltered (=) phenotype. The inlet shows the 2 main G protein switches that control rapid agonist-induced platelet adhesion: (1) RGS10/18 negatively regulates the heterotrimetic G proteins coupled to surface receptors (GPCRs) stimulated by soluble agonists such as thrombin (Thr), thromboxane A2 (TxA2), and adenosine diphosphate (ADP); (2) RASA3 negatively regulates the monomeric G protein RAP1, critical regulator of integrin-mediated adhesion. MK, megakaryocyte.
Effective hemostatic plug formation at sites of injury relies on the rapid recruitment of platelets from the bloodstream and on their near-immediate conversion from a nonadhesive to a proadhesive state. Nearly all soluble agonists generated at sites of injury stimulate platelets via heterotrimeric G protein–coupled receptors (GPCRs). Heterotrimeric G proteins consist of 3 subunits, α, β, and γ, strategically inserted in the inner leaflet of the plasma membrane to relay signals coming from the extracellular space. GPCRs engagement displaces the GDP from the Gα subunit and allows for the immediate loading of the more abundant guanosine triphosphate (GTP) onto the nucleotide-free G protein. This on-switch mechanism provides a perfect system to transduce signals on a millisecond scale required for platelet adhesion and 3-dimensional thrombus growth under shear stress conditions. However, it must be tightly controlled to avoid thrombosis or thrombocytopenia that may result from increased clearance of activated platelets.
In the middle to late 1990s, studies in yeast and Caenorhabditis elegans led to the discovery of a new family of regulators of G-protein signaling capable of reducing the amplitude and duration of GPCR signaling by increasing the rate of GTP hydrolysis and returning the G protein to the off state. Fifteen years later, the Brass group made the exciting observation that these RGS proteins may be important negative regulators of GPCR signaling also in platelets, since mice expressing a mutant Gαi subunit unable to bind RGS proteins displayed enhanced platelet function in vitro and in vivo.2 However, as they and others went on to carefully characterize the contribution of individual RGS isoforms to platelet function,3-5 the role and importance of RGS proteins were put into question. As expected, genetic ablation in mice of either one of the major RGS isoforms expressed in platelets, Rgs10 or Rgs18, shortens bleeding times as well as thrombus occlusion times in vivo, but the phenotype of the single knockouts is milder than that of the Gαi mutant and of other mouse models lacking established molecular brakes of platelets, such as the ITIM receptor G6b-B6 and the RAP1-GTPase activating protein RASA37 (see figure). One possible explanation for the mild phenotype is of course the redundancy between RGS isoforms; thus, the authors set out to investigate the phenotype of mice lacking both Rgs10 and Rgs18.
With this study, DeHelian et al demonstrate beyond further doubt that RGS10 and RGS 18 have an important and redundant role in dampening agonist-induced platelet activation and thrombus growth at sites of vascular injury. Indeed, deficiency in both RGS isoforms leads to an exaggerated platelet accumulation and frequent occlusion of injured vessels, thus supporting the idea that the mild phenotype observed in the single knockouts was just due to redundancy between the 2 isoforms. One might wonder, what is the physiological relevance of 2 related proteins that basically compensate for each other? In biological systems, redundancy provides a safety net that ensures that important signaling steps take place no matter what. Thus, it is not surprising to find redundant proteins in a crucial signaling node controlling the balance between platelet activation and platelet inhibition.
Brakes are necessary not only to limit thrombus growth but also to prevent unwanted activation of circulating platelets in the absence of an injury. For this reason, mice lacking molecular brakes of platelet activation typically display increased markers of platelet activation even in the absence of stimuli, increased platelet turnover, reduced platelet half-life, and thrombocytopenia due to increased clearance. This is not entirely the case for RGS proteins. Neither of the single knockout mice show evidence of premature platelet clearance. Rgs10−/− mice have a normal platelet count/size, and Rgs18−/− mice display a mild thrombocytopenia (∼15% reduction) caused by defects in megakaryocyte function. A phenotype becomes apparent only when both Rgs genes are deleted, again because of redundancy. The double knockout mice are characterized by circulating platelets with exposed TLT-1, a marker of secretion, an increased proportion of reticulated platelets, indicative of augmented platelet turnover, and a 40% reduction in platelet count. However, the role of RGS10 and RGS18 in limiting preactivation of circulating platelets is presumably modest, since genetic ablation of other established molecular brakes, such as RASA3, leads to more dramatic platelet count reductions.7 This different phenotype is likely due to different regulatory mechanisms. In quiescent circulating platelets, while RASA3 is always active, RGS10 and RGS18 are trapped into a complex with spinophilin that prevents them from sending tonic inhibitory signals to GPCRs in the absence of stimuli. Why are they different though? Are there other molecular brakes restraining GPCR signaling in unstimulated platelets? Or is it that RAP1 proteins are more likely than GPCRs to become randomly activated in circulation, owing to the fact that the RAP1 activator CalDAG-GEFI is extremely sensitive to intracellular calcium fluxes? More basic scientific work is needed to answer these important questions.
Finally, this study confirms that RGS18, but not RGS10, contributes to the regulation of platelet production. The authors were not able to pin down the mechanism underlying this observation. Thus, further studies are needed to dissect how this molecular brake supports normal platelet production and whether mutations in Rgs18 are associated with congenital thrombocytopenia in humans as is the case for the ITIM receptor G6b-B.8
In summary, DeHelian and colleagues identify both redundant and nonredundant roles of the 2 major isoforms of the RGS family, RGS10 and RGS18, in modulating platelet activation and platelet number. Characterization of these 2 molecular brakes may have important implications to better understand pathological conditions in which stimulatory and inhibitory signaling pathways are unbalanced.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal