

Key Points
CRISPR/Cas9-mediated T-cell-receptor knockout with anti-CD19-CAR expression enables allo-CAR–T-cell therapy.
Coexpression of endogenous TCR and CD19-CAR prolongs in vivo persistence of T cells.

Abstract
Anti-CD19 chimeric antigen receptor (CAR) T cells showed significant antileukemic activity in B-precursor acute lymphoblastic leukemia (ALL). Allogeneic, HLA-mismatched off-the-shelf third-party donors may offer ideal fitness of the effector cells, but carry the risk of graft-versus-host disease. Knockout (KO) of the endogenous T-cell receptor (TCR) in CD19-CAR-T cells may be a promising solution. Here, we induced a CRISPR/Cas9-mediated KO of the TCRβ chain in combination with a second-generation retroviral CAR transduction including a 4-1BB costimulatory domain in primary T cells. This tandem engineering led to a highly functional population of TCR-KO-CAR-T cells with strong activation (CD25, interferon γ), proliferation, and specific killing upon CD19 target recognition. TCR-KO-CAR-T cells had a balanced phenotype of central memory and effector memory T cells. KO of the endogenous TCR in T cells strongly ablated alloreactivity in comparison with TCR-expressing T cells. In a patient-derived xenograft model of childhood ALL, TCR-KO-CAR-T cells clearly controlled CD19+ leukemia burden and improved survival in vivo. However, coexpression of endogenous TCR plus CAR led to superior persistence of T cells and significantly prolonged leukemia control in vivo, confirmed by a second in vivo model using the leukemia cell line NALM6. These results point toward an essential role of the endogenous TCR for longevity of the response at the price of alloreactivity. In conclusion, anti-CD19 CAR T cells with a CRISPR/Cas9-mediated TCR-KO are promising candidates for nonmatched third-party adoptive T-cell transfer with high antileukemic functionality in the absence of alloreactivity, but long-term persistence in vivo is better in the presence of the endogenous TCR.
Introduction
Treatment with autologous anti-CD19 chimeric antigen receptor (CAR) T cells has shown high initial complete response rates in patients with relapsed and refractory acute lymphoblastic B-precursor leukemia (BCP-ALL).1,2 Nevertheless, up to 50% of pediatric patients experience relapse due to loss of CAR T cells or escape variants of leukemic blasts.3,4 Moreover, insufficient lymphocyte numbers for leukapheresis, chemotherapy pretreatment, and failure of CAR–T-cell production remain unsolved challenges in various CAR production approaches.4-6 Insufficient T-cell function has also been attributed to leukemia-induced T-cell exhaustion.7 Administration of allogeneic off-the-shelf products from healthy HLA-mismatched donors may overcome these hurdles, but carry the risk of severe graft-versus-host disease (GvHD). Current CAR–T-cell protocols use lymphodepleting regimens eliminating the host’s T-cell pool for the time of CAR–T-cell infusion and expansion, minimizing the risk of rejection of CAR T cells.3,6 T cell receptor (TCR) knockout (KO) of the CAR–T-cell product would be supposed to prevent GvHD. Two patients were reported previously who were treated with anti-CD19-CAR-T cells, engineered with transcription activator-like effector nucleases (TALENs) to KO the endogenous TCR.8 In these cases, allogeneic T cells derived from an HLA-mismatched healthy third-party donor. TALEN-mediated TCR-KO was combined with lentiviral transduction to produce TCR−/CAR+ T cells, which were administered to 2 pediatric BCP-ALL patients with good initial response rates. However, TCR+ cells from the CAR infusion expanded in vivo and induced GvHD.
The relevance of the TCR was also analyzed in a mouse model using knockin of an anti-CD19 CAR into the locus of the endogenous constant TCR α-chain (TRAC) by combination of clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) technology with adeno-associated viral transfer.9 The CAR in the TRAC locus resulted in increased CAR functionality in preclinical mouse models, but was not yet investigated in clinical phase I/II trials. Nevertheless, recently published clinical data with virus-specific T cells showed that additional TCR stimulation enhances the expansion and function of CD19-CAR-T cells.10
Here, we combine CRISPR/Cas9-mediated TCR-KO with retroviral transduction of a second-generation anti-CD19 CAR with 4-1BB-based costimulation to analyze the relevance of the endogenous TCR for functionality of TCR−/CAR+ T cells. TCR-KO-CAR-T cells exert excellent anti-CD19 activity and significantly decreased alloreactivity. However, TCR+ CAR T cells showed significantly improved persistence in vivo indicating an essential role of the endogenous TCR for sustained CAR T-cell function in vivo. Nevertheless, highly functional allogeneic TCR− CARs might be promising treatment candidates as a bridge to transplant.
Materials and methods
Isolation of peripheral blood mononuclear cells (PBMCs), T-cell activation, cell lines, flow cytometry, intracellular staining, histology, proliferation, cytotoxicity assays, CAR-enrichment, and depletion of CD3+ T cells are described in the supplemental Methods, available on the Blood Web site.
Retroviral anti-CD19 CAR transduction
For retroviral transduction, second-generation anti-CD19 CAR sequence containing FMC63,11 CD8 transmembrane and spacer domains, and 4-1BB costimulatory domain (based on patent WO2015187528A1) was cloned into pMP71 (kindly provided by Christopher Baum, Medizinische Hochschule Hannover, Hannover, Germany) via EcoRI and NotI (pMP71_CAR). A myc tag was included in the CAR for detection and purification. pMP71_CAR was transfected into 293Vec-Galv cells (kindly provided by BioVec Pharma Inc., Quebec, Canada) using TransIT-293 transfection reagent (Mirus Bio, Madison, WI) according to the supplier’s instructions. Retroviral supernatant was used for transduction of 293-Vec-RD114 cells (kindly provided by BioVec Pharma Inc.) to create a stable producer system. Retroviral supernatant of 293-Vec-RD114 cells was used to transduce primary T cells from healthy donors. Therefore, 24-well plates were coated with 2.5 μg RetroNectin reagent (Takara Bio, Kusatsu, Japan) per well at 37°C for 2 hours. Plates were blocked with 2% Albumin Fraction V (Carl Roth, Karlsruhe, Germany) in phosphate-buffered saline (PBS; Gibco; Thermo Fisher Scientific, Waltham, MA) for 30 minutes and washed with a 1:40 dilution of HEPES 1 M (Thermo Fisher Scientific) in PBS. Virus supernatant was harvested and filtered (0.45 μm). One milliliter of virus supernatant was transferred in each well of the plate and centrifuged at 3000g for 90 minutes at 32°C. Virus supernatant was discarded, and 1 × 106 T cells in 1 ml of TexMACS GMP medium (Miltenyi Biotec, Bergisch Gladbach, Germany)/2.5% human AB serum (Institute for Clinical Transfusion Medicine, Ulm, Germany) plus 12.5 ng/mL human IL-7 and IL-15, premium grade (Miltenyi Biotec) and 2 µg/mL Protamine sulfate (Sigma-Aldrich, Taufkirchen, Germany) were added. Plates were centrifuged for 10 minutes at 450g at 32°C. Transduction was performed on day 2 after T-cell activation.
CRISPR/Cas9-mediated TCR KO
CRISPR/Cas9-mediated TCR KO was performed 1 day after CAR transduction. The guide RNA (gRNA) targeting the TCR constant β-chain was published previously.12,13 For CRISPR/Cas9-mediated TCR KO, Alt-R CRISPR-Cas9 transactivating CRISPR RNA and Alt-R CRISPR-Cas9 CRISPR RNA (both from Integrated DNA Technologies, Coralville, IA) were mixed 1:1 and heated 5 minutes at 95°C. Alt-R S.p. Cas9 Nuclease 3NLS (Integrated DNA Technologies) was mixed with the gRNA complex and Alt-R Cas9 Electroporation Enhancer (Integrated DNA Technologies) and incubated 15 minutes at room temperature. For electroporation, 1 M buffer14 was used on a Nucleofector 2b device according to the manufacturer’s instructions (Lonza, Basel, Switzerland). After electroporation, T cells were immediately transferred to fresh medium. T cells treated with a nonbinding gRNA were used as control (Integrated DNA Technologies). T cells were further expanded as described in the supplemental Methods.
Alloreactivity assay
PBMCs were isolated from 6 different healthy donors, irradiated with 20 Gy, and pooled. To distinguish between TCR KO T cells and donor-derived allogeneic PBMCs (allo-PBMCs), T cells were labeled with CellTrace CFSE Cell Proliferation Kit and PBMCs were labeled with CellTrace Violet Cell Proliferation Kit (both Thermo Fisher Scientific). After coculturing the cells at a 1:5 effector-to-target (E:T) ratio for 1 to 5 days, TCR-KO vs wildtype T cells were analyzed for their activation marker profile (CD69, CD137, CD25) and their proliferation by flow cytometry.
In vivo experiments
All animal trials were performed in accordance with the current ethical standards of the official committee on animal experimentation (written approval by Regierung von Oberbayern; ROB-55.2Vet-2532.Vet_02-16-7). Mice were maintained under specific pathogen-free conditions, had free access to food and water, and were housed with a 12-hour light/dark cycle and constant temperature. The leukemia cell line NALM6 and ALL-265 patient-derived xenograft (PDX) cells15 were genetically modified by lentiviral transduction to express enhanced firefly luciferase and enhanced GFP (eGFP) as selection marker (see Addgene vector 104834).16 A total of 1 × 105 luciferase-positive NALM6 or 2 × 106 BCP-ALL PDX cells were transplanted IV into NOD-scid IL2Rγnull (NSG) mice (The Jackson Laboratory, Bar Harbor, ME). Three days after injection, CAR T cells were thawed and counted, and 2 × 107 cells in 200 µl of PBS were injected IV into mice. This high number of T cells was chosen to increase the risk of GvHD. Early onset of GvHD clarifies differences of alloreactivity and tumor growth control between TCR+ CAR T cells and TRBC KO CAR T cells. Leukemia burden was monitored once or twice per week by bioluminescence in vivo imaging (BLI) as described previously.15,17 Furthermore, peripheral blood was analyzed regularly to detect presence of human T and CAR T cells. Mice were monitored daily for signs of GvHD like weight loss, hair loss, altered posture, or reduced mobility. When BLI reached values above 1 × 1010 P/sec, or if mice showed clinical signs of illness or GvHD, mice were euthanized and blood was analyzed for presence of NALM6 or PDX cells and human T/CAR T cells.
Statistics
Statistical analyses were performed using GraphPad Prism 7. A 2-tailed paired Student t-test was performed to analyze the statistical significance between experimental conditions of at least 3 independent experiments. When more than 2 groups were compared with each other, statistical significance was tested with a one-way ANOVA. Overall survival of mice was calculated using the Kaplan-Meier method. A two-tailed Mann-Whitney U test was used to compare presence of T cells in the peripheral blood of mice. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Results
CAR T cells with KO of the TCRβ chain show comparable transduction rates and expansion compared to conventional CAR T cells
T cells were isolated, activated (CD3/CD28 stimulation; Figure 1A), and retrovirally transduced with a second-generation CAR (Figure 1B). One day after transduction, CRISPR/Cas9-mediated TCR-KO was performed by electroporation of the ribonucleoprotein complex including a gRNA targeting the TCRβ chain (TRBC). Nonelectroporated conventional CARs (RV19BB_TCR+), CAR T cells electroporated with Cas9 and a nonbinding gRNA (RV19BBCRTCR+), and untransduced, nonelectroporated T cells served as comparator for CAR T cells with KO of the TCRβ chain (RV19BBCRTRBC−). The gRNA targeting the TCRβ chain was published before and showed high on-target and low off-target effects.18 Because the TCR β constant region comprises 2 different genes, TRBC1 and TRBC2, we made sure that the selected gRNA targets both TRBC loci. RV19BB_TCR+, RV19BBCRTCR+, and RV19BBCRTRBC− CARs showed comparable mean transduction rates of 36.6%, 38.3%, and 39.6%, respectively (Figure 1C). RV19BBCRTRBC− CAR T cells reached a mean TCR-KO rate of 78.2%, determined by flow cytometry (Figure 1D). A total of 32.2% of all T cells had both a CAR on the surface as well as a TCR-KO after expansion (Figure 1E). Exemplary flow cytometry plots to determine transduction and TCR-KO rates are shown in Figure 1F. Defective TCR assembly upon KO of the TCRβ chain is confirmed by simultaneous CD3 downregulation (Figure 1G). After magnetic CAR enrichment (c-myc tag) and CD3 depletion, a high purity of TCR−/CAR+ T cells was achieved (supplemental Figure 1A). Untransduced T cells and RV19BB_TCR+ CARs showed an expansion >120-fold (175-fold and 124-fold, respectively), whereas RV19BBCRTCR+ and RV19BBCRTRBC− CARs showed slightly reduced proliferative capacity (77-fold and 89-fold, respectively; Figure 1H). Expansion impairment was seen in RV19BBCRTCR+ and RV19BBCRTRBC− CARs, meaning that this effect was mediated by the electroporation process itself rather than by loss of the TCR.
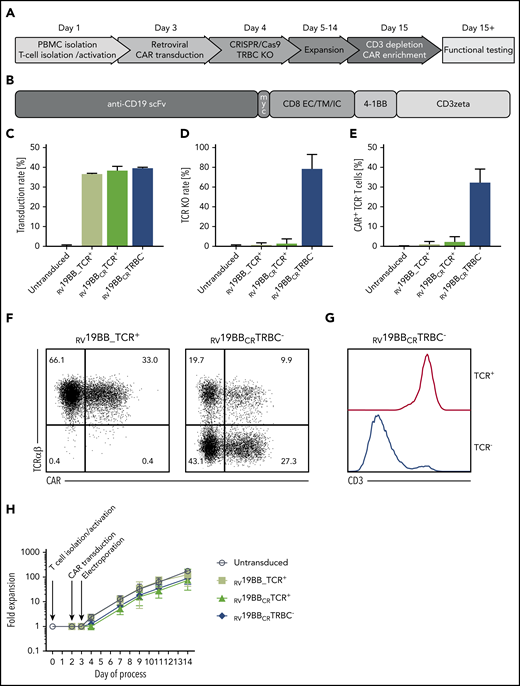
Generation of TCR KO CAR T cells. (A) Schematic overview of the time points for retroviral transduction, CRISPR/Cas9-mediated TCR KO, duration of cultivation, and purification of the final CAR–T-cell product. (B) Structure of the CAR construct: the second-generation CAR consists of the anti-CD19 scFv region, followed by the myc tag, CD8 extracellular (EC), transmembrane (TM), and intracellular (IC) domain, as well as a costimulatory 4-1BB domain and T-cell–activating CD3ζ chain. (C) A mean CAR transduction rate of >35% could be reached for the RV19BB_TCR+, RV19BBCRTCR+, and RV19BBCRTRBC− CARs as assessed by flow cytometry (3 independent experiments). (D) The mean TCR KO rate within CD4+CD8+ cells was 78.2%, and (E) the proportion of cells expressing the CAR and lacking the TCR reached around 32.2% (3 independent experiments). (F) Exemplary flow cytometry plots to determine transduction rate and TCR KO efficacy, and (G) correlation of CD3 and TCR expression in TCR+ and TCR− T cells within RV19BBCRTRBC− CARs is shown in this histogram. (H) Fold expansion of the untransduced T cells, RV19BB_TCR+, RV19BBCRTCR+, and RV19BBCRTRBC− CARs after T-cell isolation and activation, transduction, and electroporation (n ≥ 4). PB, peripheral blood.
Generation of TCR KO CAR T cells. (A) Schematic overview of the time points for retroviral transduction, CRISPR/Cas9-mediated TCR KO, duration of cultivation, and purification of the final CAR–T-cell product. (B) Structure of the CAR construct: the second-generation CAR consists of the anti-CD19 scFv region, followed by the myc tag, CD8 extracellular (EC), transmembrane (TM), and intracellular (IC) domain, as well as a costimulatory 4-1BB domain and T-cell–activating CD3ζ chain. (C) A mean CAR transduction rate of >35% could be reached for the RV19BB_TCR+, RV19BBCRTCR+, and RV19BBCRTRBC− CARs as assessed by flow cytometry (3 independent experiments). (D) The mean TCR KO rate within CD4+CD8+ cells was 78.2%, and (E) the proportion of cells expressing the CAR and lacking the TCR reached around 32.2% (3 independent experiments). (F) Exemplary flow cytometry plots to determine transduction rate and TCR KO efficacy, and (G) correlation of CD3 and TCR expression in TCR+ and TCR− T cells within RV19BBCRTRBC− CARs is shown in this histogram. (H) Fold expansion of the untransduced T cells, RV19BB_TCR+, RV19BBCRTCR+, and RV19BBCRTRBC− CARs after T-cell isolation and activation, transduction, and electroporation (n ≥ 4). PB, peripheral blood.
In vitro CAR–T-cell function is independent of presence or absence of endogenous TCR
T-cell characteristics with or without TCR were analyzed (CD62L/CD95/CD45RO expression; Figure 2A).19 After expansion (14 days IL-7/IL-15), no naïve T cells and only a minor fraction (<2.2%) of terminally differentiated effector T cells (Teff) were detectable in untransduced RV19BB_TCR+, RV19BBCRTCR+ and RV19BBCRTRBC− CAR T cells. Thus, the final T-cell product consisted mainly of central memory (Tcm) and effector memory (Tem) T cells. T-cell subpopulations were distributed equally between RV19BB_TCR+ CAR T cells, with a mean of 50.6% Tcm and 47.8% Tem, and the RV19BBCRTRBC− CARs, with a mean of 47.1% Tcm and 46.0% Tem cells. Expression of costimulatory and coinhibitory molecules as well as exhaustion markers 2B4, LAG-3, and PD-1 was analyzed by flow cytometry at the end of expansion phase (Figure 2B). RV19BBCRTRBC− CAR T cells shared a comparable profile of costimulatory and inhibitory molecules with RV19BB_TCR+ CAR T cells. RV19BB_TCR+ and RV19BBCRTRBC− CARs showed high surface expression levels of OX40 (52.4% vs 46.9%) and CD28 (88.2% vs 89.0%), whereas 4-1BB was expressed to a very low amount at the end of the expansion protocol. Both RV19BB_TCR+ and RV19BBCRTRBC− CARs displayed low expression of coinhibitory molecules like PD-1 (5.5% vs 3.2%), BTLA (2.7% vs 0.6%), CTLA-4 (0.4% vs 0.2%), TIGIT (12.4% vs 10.6%), and VISTA (0.1% vs 0.1%). Only TIM-3 was highly expressed on RV19BB_TCR+ as well as even slightly increased on RV19BBCRTRBC− CAR T cells (80.8% vs 85.8%; P = .0222). Besides that, the TCR-KO on CAR T cells had no effect on the expression of typical exhaustion markers like 2B4 (11.1% vs 15.5%), LAG-3 (7.1% vs 6.4%), and PD-1 (5.5% vs 3.2%). At the end of the expansion protocol, different T-cell subsets were present in the final product, but no monocytes, B cells or natural killer (NK) cells (Figure 2C). RV19BBCRTRBC− CARs and RV19BB_TCR+ CARs showed a similar cellular composition of CD8+ T cells (47.5% vs 37.1%) and NKT cells (17.4% vs 13.5%), with minor differences in the CD4+ T-cell population (32.6% vs 44.8%, P = .0222).
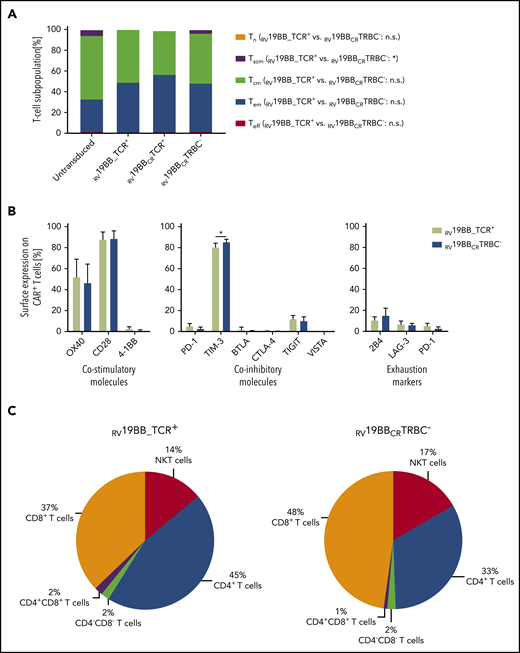
Cellular characteristics of the final CAR–T-cell product. (A) The final product of CAR-expressing T cells showed mainly Tcm and Tem T cells regardless of TCR expression after expansion (n = 3). (B) Surface expression profile of several costimulatory (OX40, CD28, 4-1BB) and coinhibitory molecules (PD-1, TIM-3, BTLA, CTLA-4, TIGIT, VISTA) as well as commonly used exhaustion markers (2B4, LAG-3, PD-1) was determined by flow cytometry and compared between CAR T cells with or without TCR KO (n = 3). (C) Cellular composition of CAR T cells was determined on day 14 (n = 4). A 2-tailed paired Student t test was performed to determine statistical significance. Tcm, CD62L+, CD45RO+, CD95+; Tem, CD62L−, CD45RO+, CD95+; Teff, CD62L−, CD45RO−, CD95+; Tn, CD62L+, CD45RO−, CD95−; Tscm, CD62L+, CD45RO−, CD95+. n.s., not significant; Tn, naïve T cells; Tscm, stem cell-like memory T cells.
Cellular characteristics of the final CAR–T-cell product. (A) The final product of CAR-expressing T cells showed mainly Tcm and Tem T cells regardless of TCR expression after expansion (n = 3). (B) Surface expression profile of several costimulatory (OX40, CD28, 4-1BB) and coinhibitory molecules (PD-1, TIM-3, BTLA, CTLA-4, TIGIT, VISTA) as well as commonly used exhaustion markers (2B4, LAG-3, PD-1) was determined by flow cytometry and compared between CAR T cells with or without TCR KO (n = 3). (C) Cellular composition of CAR T cells was determined on day 14 (n = 4). A 2-tailed paired Student t test was performed to determine statistical significance. Tcm, CD62L+, CD45RO+, CD95+; Tem, CD62L−, CD45RO+, CD95+; Teff, CD62L−, CD45RO−, CD95+; Tn, CD62L+, CD45RO−, CD95−; Tscm, CD62L+, CD45RO−, CD95+. n.s., not significant; Tn, naïve T cells; Tscm, stem cell-like memory T cells.
CAR T cells with KO of the TCRβ chain are highly functional in vitro and prevent alloreactivity
Activation potential of TCR-KO-CAR-T cells was tested (Figure 3A). Untransduced T cells, RV19BB_TCR+, RV19BBCRTCR+, and RV19BBCRTRBC− CARs were cocultured with a CD19+ target cell line for 24 hours and then stained for the activation markers CD137 and CD25. The baseline surface expression of both markers was very low, ranging from 0.8% to 6.0% for CD137 and from 1.2% to 4.2% for CD25. Upon antigen contact, the CAR-expressing T cells showed a significant upregulation of CD137 and CD25 compared to untransduced T cells. RV19BBCRTRBC− CARs displayed a similar target-specific activation compared to RV19BB_TCR+ CAR T cells with a mean CD137 expression of 65.0% vs 66.3% and a mean CD25 expression of 95.2% vs 94.7%. To determine the lytic potential of RV19BBCRTRBC− CAR T cells, cytokine secretion after 24 hours of coculturing with CD19+ target cells was analyzed (Figure 3B). Notably, interindividual variability in cytokine secretion was compensated by analysis of 4 different individual donors. Without antigen contact, untransduced T cells, RV19BB_TCR+, RV19BBCRTCR+, and RV19BBCRTRBC− CARs showed <0.5% of interferon γ (IFNγ)–positive cells. After coculturing with target cells, RV19BB_TCR+, RV19BBCRTCR+, and RV19BBCRTRBC− CARs showed a significantly increased percentage of IFNγ-positive cells ranging from 5.3% to 30.4% of RV19BB_TCR+ CARs, 8.8% to 22.4% of RV19BBCRTCR+ CARs, and 10.9% to 23.2% of RV19BBCRTRBC− CARs. A similar pattern was observed for tumor necrosis factor α (TNFα) secretion upon stimulation. RV19BB_TCR+ CAR T cells as well as RV19BBCRTRBC− CARs showed a significant upregulation of TNFα-positive cells upon antigen stimulation (5.5% up to 12.3% vs 4.3% up to 12.1%). Therefore, loss of the TCR on RV19BBCRTRBC− CARs showed no disadvantage regarding secretion of IFNγ and TNFα compared to RV19BB_TCR+ CARs. The proliferative characteristics of RV19BBCRTRBC− CARs showed comparable results to RV19BB_TCR+ CARs after 72 hours of antigen contact (Figure 3C). More than 97.6% of RV19BB_TCR+, RV19BBCRTCR+ and RV19BBCRTRBC− CARs proliferated. To analyze the cytotoxic capacity, the different CAR approaches were cocultured with CD19+ target cells for 48 hours (Figure 3D). RV19BB_TCR+, RV19BBCRTCR+, and RV19BBCRTRBC− CARs showed high anti-CD19 cytotoxicity at various E:T ratios independent of TCR expression. At an E:T ratio of 1:1, RV19BB_TCR+, RV19BBCRTCR+, and RV19BBCRTRBC− CARs showed a mean killing of 87.6%, 86.1%, and 84.6%, respectively. Even at a very low E:T ratio of 0.04:1, >36% of the CD19-expressing target cells were killed by the CAR T cells, indicating strong efficacy even at low T-cell concentrations.
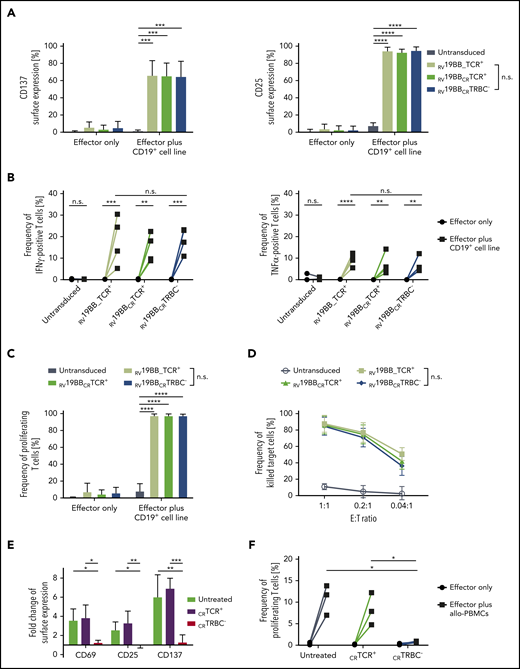
Functionality of RV19BBCRTRBC− CAR T cells and alloreactive potential of TCR+ T cells in vitro. (A-D) The functionality of RV19BBCRTRBC− CAR T cells was compared to RV19BB_TCR+, RV19BBCRTCR+ CARs and untransduced T cells in different functionality assays. (A) Twenty-four hours after coculturing the CAR T cells with CD19+ target cells in a 1:1 E:T ratio, the cells were harvested and analyzed for expression of activation markers CD137 and CD25 (n = 6). (B) Intracellular staining of IFNγ and TNFα was performed after 24 hours of coculturing effector cells with CD19+ cells at an E:T of 1:1 (n = 4). (C) CFSE labeled T cells were used to analyze the frequency of proliferating effector cells after contact with CD19+ target cells for 72 hours (n = 3). (D) CD19+ target-cell killing was determined by flow cytometry after target cells were cocultured for 48 hours with CAR T cells in different E:T ratios (n = 4). (E-F) Untransduced, CRTCR+, CRTRBC− T cells were cocultured with allo-PBMCs pooled from 6 different donors and cocultured at an E:T ratio of 1:5. (E) After 48 hours of coculture, T cells were analyzed for surface expression of the activation markers CD69, CD25, and CD137 (n = 3). (F) Percentage of proliferating T cells after contact with allo-PBMCs was analyzed after 5 days (n = 3). A 2-tailed paired Student t test or 1-way ANOVA was performed to determine statistical significance.
Functionality of RV19BBCRTRBC− CAR T cells and alloreactive potential of TCR+ T cells in vitro. (A-D) The functionality of RV19BBCRTRBC− CAR T cells was compared to RV19BB_TCR+, RV19BBCRTCR+ CARs and untransduced T cells in different functionality assays. (A) Twenty-four hours after coculturing the CAR T cells with CD19+ target cells in a 1:1 E:T ratio, the cells were harvested and analyzed for expression of activation markers CD137 and CD25 (n = 6). (B) Intracellular staining of IFNγ and TNFα was performed after 24 hours of coculturing effector cells with CD19+ cells at an E:T of 1:1 (n = 4). (C) CFSE labeled T cells were used to analyze the frequency of proliferating effector cells after contact with CD19+ target cells for 72 hours (n = 3). (D) CD19+ target-cell killing was determined by flow cytometry after target cells were cocultured for 48 hours with CAR T cells in different E:T ratios (n = 4). (E-F) Untransduced, CRTCR+, CRTRBC− T cells were cocultured with allo-PBMCs pooled from 6 different donors and cocultured at an E:T ratio of 1:5. (E) After 48 hours of coculture, T cells were analyzed for surface expression of the activation markers CD69, CD25, and CD137 (n = 3). (F) Percentage of proliferating T cells after contact with allo-PBMCs was analyzed after 5 days (n = 3). A 2-tailed paired Student t test or 1-way ANOVA was performed to determine statistical significance.
For analysis of alloreactivity in vitro, CD19 expressing B cells are necessary for sufficient induction of alloreactivity and anti-CD19-reactivity becomes indistinguishable from alloreactivity for CAR T cells in a mixed lymphocyte reaction. Instead, wildtype T cells with (CRTCR−) or without a TCR KO (CRTCR+) were analyzed for alloreactivity. Untreated T cells, CRTCR+, and CRTCR− T cells were cocultured with irradiated PBMCs pooled from 6 different donors. After 48 hours of coculture, expression of activation markers CD69, CD25, and CD137 was analyzed (Figure 3E). Untreated T cells and CRTCR+ showed a significant upregulation of CD69 (3.6-fold and 3.9-fold, respectively), CD25 (2.6-fold and 3.3-fold, respectively), and CD137 (6.1-fold and 7.0-fold, respectively) upon contact with allo-PBMCs compared to CRTCR− T cells, which showed no response to allo-PBMCs (CD69, 1.3-fold; CD25, 0.9-fold; CD137, 1.3-fold). A similar proliferative response was seen 5 days after allo-PBMC stimulation (Figure 3F). Untreated T cells and CRTCR+ T cells showed elevated levels of proliferating cells ranging from 7.0% to 13.8% and 4.8% to 12.2% upon stimulation, respectively, whereas CRTCR− T cells showed almost no response after contact with allo-PBMCs (0.7% to 0.9% proliferating cells), pointing toward significantly reduced alloreactivity of TCR-deficient T cells.
Patient derived xenografts of CD19+ childhood acute lymphoblastic leukemia (ALL) induce sufficient activation and target-cell killing of anti-CD19-CAR-T cells
To investigate whether RV19BBCRTRBC− CAR T cells can eliminate allogeneic, patient-derived tumor cells in vitro, PDX leukemic cells were expanded in a murine model. Leukemic cells from a pediatric BCP-ALL patient were transferred to NSG mice, expanded over several passages and genetically modified to express enhanced firefly luciferase and eGFP as selection marker.16,17 Passage 3 and passage 5 PDX cells were extracted from the bone marrow of NSG mice, and expression of CD19 was confirmed by flow cytometry (supplemental Figure 1B). To determine the killing capacity of CAR T cells, in vitro cytotoxicity assays at a 0.2:1 E:T ratio were performed (Figure 4A). RV19BB_TCR+, RV19BBCRTCR+, and RV19BBCRTRBC− CARs showed a significantly increased killing rate (60.5%, 63.1%, and 64.3%, respectively) of ALL-265 PDX cells compared to untransduced T cells (0.2%). Cytotoxicity of RV19BBCRTRBC− CAR T cells was comparable to conventional CAR T cells. We next investigated the proliferative capacity of RV19BBCRTRBC− CAR T cells upon contact with ALL-265 PDX cells for 72 hours (Figure 4B). RV19BB_TCR+, RV19BBCRTCR+, and RV19BBCRTRBC− CARs proliferated upon contact with ALL-265 PDX cells ranging from 14.2% to 68.2%, 24.1% to 55.6%, and 17.0% to 59.9% of proliferating cells, respectively.

CAR T cells against PDX cells in vitro and long-lasting leukemia control in vivo. (A) Untransduced T cells and CAR T cells were cocultured with CD19+ ALL-265 PDX cells at an E:T ratio of 0.2:1. Cytotoxicity was investigated 48 hours later (n = 3). (B) Untransduced T cells, RV19BB_TCR+, RV19BBCRTCR+, and RV19BBCRTRBC− CARs were cocultured with CD19+ ALL-265 PDX cells at an E:T ratio of 1:1. Proliferation was analyzed after 72 hours by flow cytometry (n = 3). A 2-tailed paired Student t test or 1-way ANOVA was performed to determine statistical significance. (C) NSG mice were injected with ALL-265 PDX cells, followed by IV T-cell injection (2 × 107 cells) of RV19BBCRTCR+ CAR T cells or RV19BBCRTRBC− CAR T cells or PBS 3 days after. (D-E) At indicated time points after T-cell injection, PDX leukemia burden was monitored by BLI. (D) Bioluminescence pictures and (E) quantification of leukemia burden are shown for mice suffering from ALL-265 PDX-cell–induced leukemia and treated with PBS (n = 3), RV19BBCRTCR+ (n = 5), or RV19BBCRTRBC− (n = 4; see also supplemental figures). (F) Kaplan-Meier analysis of survival of mice treated with PDX cells. A 2-tailed paired Student t test was performed to determine statistical significance.
CAR T cells against PDX cells in vitro and long-lasting leukemia control in vivo. (A) Untransduced T cells and CAR T cells were cocultured with CD19+ ALL-265 PDX cells at an E:T ratio of 0.2:1. Cytotoxicity was investigated 48 hours later (n = 3). (B) Untransduced T cells, RV19BB_TCR+, RV19BBCRTCR+, and RV19BBCRTRBC− CARs were cocultured with CD19+ ALL-265 PDX cells at an E:T ratio of 1:1. Proliferation was analyzed after 72 hours by flow cytometry (n = 3). A 2-tailed paired Student t test or 1-way ANOVA was performed to determine statistical significance. (C) NSG mice were injected with ALL-265 PDX cells, followed by IV T-cell injection (2 × 107 cells) of RV19BBCRTCR+ CAR T cells or RV19BBCRTRBC− CAR T cells or PBS 3 days after. (D-E) At indicated time points after T-cell injection, PDX leukemia burden was monitored by BLI. (D) Bioluminescence pictures and (E) quantification of leukemia burden are shown for mice suffering from ALL-265 PDX-cell–induced leukemia and treated with PBS (n = 3), RV19BBCRTCR+ (n = 5), or RV19BBCRTRBC− (n = 4; see also supplemental figures). (F) Kaplan-Meier analysis of survival of mice treated with PDX cells. A 2-tailed paired Student t test was performed to determine statistical significance.
To analyze CAR–T-cell functionality in vivo, ALL-265 PDX cells were injected IV into NSG mice and followed by T-cell injection 3 days later (Figure 4C). Mice were monitored for leukemia burden by bioluminescence. RV19BBCRTCR+ and RV19BBCRTRBC− CARs were able to immediately control growth of PDX ALL cells within 4 days below detection threshold (<1 × 106 P/sec; Figure 4D-E; supplemental Figure 2A). Although PBS control mice had to be euthanized between day 36 and 43 after T-cell injection because of end-stage leukemia, RV19BBCRTRBC− CARs increased survival to at least 76 days (Figure 4F). In contrast, mice treated with RV19BBCRTCR+ CARs completely controlled leukemia burden, but 4 mice had to be euthanized between day 38 and 61 because of clinical signs of GvHD, such as skin rash and ruffled fur (pictures not shown) and an eye infection.
Presence of endogenous TCR significantly improves persistence of CAR T cells in vivo
To further analyze CAR–T-cell functionality and the relevance of the endogenous TCR in vivo, we used the more aggressive NALM6 in vivo model and injected those IV in NSG mice, followed by T-cell injection 3 days later. Mice treated with untransduced T cells showed a fast increase in leukemia burden and had to be euthanized 22 days after T-cell injection (Figure 5A-C). In contrast, mice receiving RV19BBCRTRBC− or RV19BBCRTCR+ CARs showed a clear and comparable control of leukemia growth until day 14 post T-cell injection (average, 2.4 × 107 P/sec vs 1.8 × 107 P/sec; Figure 5A-B; supplemental Figure 2B). After day 14, treatment with RV19BBCRTCR+ CARs led to an improved prevention of leukemia regrowth compared to RV19BBCRTRBC− CAR T cells. Moreover, administration of RV19BBCRTCR+ CAR T cells resulted in prolonged survival rates compared to RV19BBCRTRBC− CAR–T-cell treatment (Figure 5C).
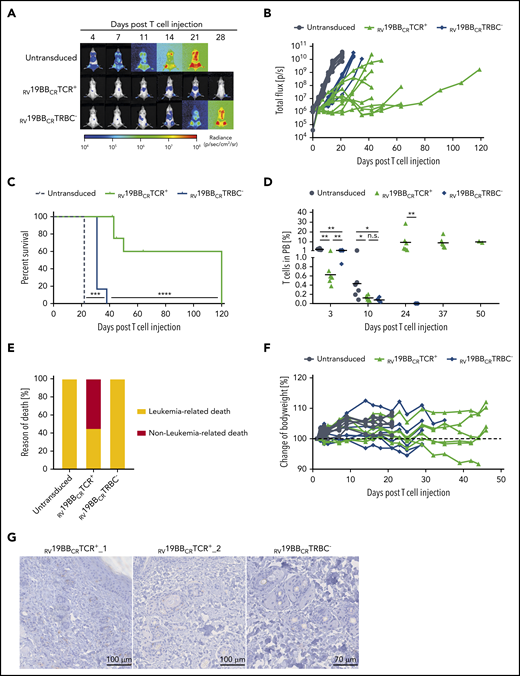
Functionality of RV19BBCRTCR+ and RV19BBCRTRBC− CAR T cells in vivo. (A-H) NSG mice were injected IV with 1 × 105 NALM6, followed by an IV T-cell injection (2 × 107 cells) of untransduced T cells (n = 6), RV19BBCRTCR+ (n = 9), and RV19BBCRTRBC− CAR T cells (n = 6) 3 days after. (A,B) At the indicated time points after T-cell injection, NALM6 leukemia burden was monitored by BLI. (A) Exemplary bioluminescence pictures are shown, as well as (B) changes in NALM6 leukemia load for each mouse. (C) Kaplan-Meier analysis of survival of mice treated with 1 × 105 NALM6 cells (n = 6 per group) is shown. A log-rank Mantel-Cox test was used to test for statistical significance. (D) T-cell persistence in peripheral blood was measured by flow cytometry. A 2-tailed Mann-Whitney U test was performed to determine statistical significance. (E) Overview of the incidence of leukemia- or nonleukemia-related death in mice. (F) Body weight was monitored at several time points after T-cell injection. (G) Exemplary micrographs of skin tissue stained for CC3 by IHC are shown for mice treated with RV19BBCRTCR+ or RV19BBCRTRBC− CARs.
Functionality of RV19BBCRTCR+ and RV19BBCRTRBC− CAR T cells in vivo. (A-H) NSG mice were injected IV with 1 × 105 NALM6, followed by an IV T-cell injection (2 × 107 cells) of untransduced T cells (n = 6), RV19BBCRTCR+ (n = 9), and RV19BBCRTRBC− CAR T cells (n = 6) 3 days after. (A,B) At the indicated time points after T-cell injection, NALM6 leukemia burden was monitored by BLI. (A) Exemplary bioluminescence pictures are shown, as well as (B) changes in NALM6 leukemia load for each mouse. (C) Kaplan-Meier analysis of survival of mice treated with 1 × 105 NALM6 cells (n = 6 per group) is shown. A log-rank Mantel-Cox test was used to test for statistical significance. (D) T-cell persistence in peripheral blood was measured by flow cytometry. A 2-tailed Mann-Whitney U test was performed to determine statistical significance. (E) Overview of the incidence of leukemia- or nonleukemia-related death in mice. (F) Body weight was monitored at several time points after T-cell injection. (G) Exemplary micrographs of skin tissue stained for CC3 by IHC are shown for mice treated with RV19BBCRTCR+ or RV19BBCRTRBC− CARs.
Peripheral blood was regularly taken to analyze T-cell persistence (Figure 5D). Untransduced T cells, RV19BBCRTCR+ CARs, and RV19BBCRTRBC− CAR T cells showed differences in engraftment 3 days after injection (mean of 2.4% vs 0.6% vs 1.3%, respectively) and a reduction by day 10, with comparable T-cell amounts in mice treated with RV19BBCRTCR+ CARs and RV19BBCRTRBC− CAR T cells (0.1% vs 0.1%, respectively). The majority of analyzed T cells were CD4+, independent of time point and genotype (supplemental Figure 2C). In the further course, T cells showed strong expansion in the RV19BBCRTCR+ CAR–T-cell group, which was stable from day 24 on, whereas no T cells were detectable anymore in mice with RV19BBCRTRBC− CAR T cell treatment after day 24. Although all mice receiving untransduced T cells or RV19BBCRTRBC− CAR T cells had to be euthanized due to high leukemia burden (6 out of 6 mice each), high leukemia burden only occurred in 44.4% of the RV19BBCRTCR+ CAR T cell group (4 out of 9 mice; Figure 5E). Instead, 4 mice were euthanized as they showed signs of GvHD in absence of high tumor burden upon day 45 and 64 after T-cell injection, with slight weight loss (Figure 5F), skin rash, and ruffled fur (pictures not shown), and 1 mouse died during anesthesia. Cleaved caspase 3 (CC3) staining of skin tissue was performed to monitor further signs of GvHD (Figure 5G). In some mice treated with TCR+ CARs (RV19BBCRTCR+_1, RV19BBCRTCR+_2), several apoptotic cells surrounding hair follicles were detectable, whereas leukemia load was comparably low at the time point of euthanization (data not shown). However, apoptotic cells were not detectable in all mice that had to be euthanized due to weight loss or clinical signs of discomfort.
Taken together, TCR KO does not impair antileukemia activity of CAR T cells in the xenograft mouse model in vivo. T-cell persistence depends on TCR-mediated alloreactivity, leading to long-term leukemia control with relevant risk of GvHD.
Discussion
In this study, we address the hurdles for off-the-shelf CD19-CAR-T cells against ALL using allogeneic donors. Relevance of the endogenous TCR is investigated using CRISPR/Cas9 KO of the constant TCRβ chain. TCR−/CAR+ T cells were highly functional and showed no alloreactivity in vitro and in vivo compared to CAR T cells with an endogenous TCR. CAR T cells with or without a TCR demonstrated both improved survival rates in an ALL patient-derived xenograft mouse model as well as in a NALM6 leukemia bearing xenogeneic mouse model in comparison with mice treated with untransduced T cells. However, only in the presence of endogenous TCRs, the CAR T cells showed prolonged and sustained persistence in vivo.
The TCR is essential for T-cell activation upon recognition of pathogens or tumor cells. Both chains, the α- and β-chain, are required for assembly of a TCRαβ and hence both KO strategies targeting the α- or β-chain result in complete absence of the TCR.12,18,20 Pilot experiments with TRAC and TRBC KO, resulted in slightly higher TCR KO efficacy when targeting the TRBC locus (data not shown). Therefore, a TRBC-specific gRNA was used for further evaluation in this study. For this TRBC-specific gRNA, high on-target and low off-target events were previously confirmed.18 Besides that, most of the trials investigated CARs with a TCR KO are targeting the α-chain, eg, with zinc-finger nucleases, TALEN or CRISPR/Cas9, so additional data for targeting the β-chain is of great interest.8,9,20 Using TCR-deficient T cells, potentially impaired cellular function has to be excluded. Characteristics and functionality of TCR-deficient CARs were well preserved compared to conventional second-generation CAR T cells in vitro with a balanced CD4/CD8 ratio and a promising phenotype of mainly Tcm and Tem, that are known to have high proliferative capacity as well as functionality in vivo.1,21 Both TCR− as well as TCR+ CAR T cells showed a comparable fraction of NKT cells in the final cell product. In a previously published study,5 we could show that this is not mediated by the T-cell–isolation method, because both CD4/CD8 and an untouched T cell enrichment include subpopulations of NKT cells.5 TCR−/CAR+ T cells highly expressed costimulatory molecules, which enhance CAR–T-cell functionality in the presence of their ligands. TCR-deficient CAR T cells did not express relevant numbers of coinhibitory molecules. Only TIM-3 was highly expressed on the final CAR–T-cell product, which is most likely mediated by anti-CD3/CD28-stimulation and IL-7/IL-15–supplemented cell culture media and could thus be a sign of activation, as shown before.22 Based on recent data, the minor differences in expression of TIM-3 on TCR-deficient CARs compared to conventional CARs, are not expected to be of functional relevance.23 Furthermore, exhaustion markers were analyzed and showed almost no surface expression, underlining the promising cellular characteristics of the TCR-deficient CAR T cells. Although conventional CAR T cells showed high alloreactive potential, no alloreactivity of TCR-KO T cells was seen in vitro and in vivo.
In the presence of CD19+ target cells in vitro, TCR-deficient CAR T cells displayed high functionality, with similar upregulation of activation markers, cytokine secretion, proliferative capacity and cytotoxicity compared to conventional CAR T cells. This confirms that early CAR–T-cell activation is not dependent on endogenous TCR signals, but rather is mediated by the CAR-intrinsic CD3ζ and 4-1BB signaling domains. This was also supported through data from our in vivo model, where leukemia-bearing mice treated with RV19BBCRTRBC− or RV19BBCRTCR+ CARs showed comparable initial control of NALM6 and ALL-265 PDX leukemia burden. Durable persistence of CAR T cells in clinical studies has been described in patients with successful leukemia control,24 emphasizing that persistence of CAR T cells is essential to control leukemia in the long term. Due to lack of persistence of RV19BBCRTRBC− CAR T cells, mice treated with those T cells showed minor long-term leukemia control than mice receiving RV19BBCRTCR+ CAR T cells. An association of CAR–T-cell persistence with alloimmune or autoimmune T-cell activation through the TCR has not been described until now. However, TCR engagement negatively affected CD8 but not CD4 CAR–T-cell expansion and leukemic clearance in an immunocompetent syngeneic murine model.25 Therefore, the endogenous TCR might play a role in activation or stabilization of the T cell and thereby prolong in vivo persistence and expansion. Specificity of these responses might be infectious or allogeneic (xenogeneic in case of mouse tissue), since several studies have shown induction of GvHD when human T cells are injected into NSG mice.26-28 Furthermore, in patients treated with HLA-mismatched, allogeneic, TALEN-engineered, TCR-deficient CAR T cells, only a small fraction of contaminating TCR+ CAR T cells persisted and induced GvHD.8 Lapteva et al10 could show that additional stimulation through a virus-specific TCR on CAR T cells led to enhanced proliferation and functionality in patients, underlining the important role of the TCR. Another study could show that CARs with CD3ζ transmembrane domains dimerize with endogenous TCR/CD3 complexes, probably resulting in phosphorylation of endogenous immunoreceptor tyrosine-based activation motifs (ITAMs) and thereby strengthening CAR activation.29 Constitutive tonic signaling in the absence of ligand is an increasingly recognized complication of engineered T cells and can be a cause of poor antitumor efficacy, impaired survival, and reduced persistence in vivo.30 This constitutive or chronic cell signaling may have a substantial deleterious impact on CAR–T-cell effector function and survival and may lead to a significant disparity between in vitro cytolytic capacity and in vivo antitumor efficacy. In our in vivo data, long-term control of ALL >120 days through an RV19BBCRTCR+ CAR excludes predominance of this deleterious signaling and demonstrates capacity for sustained in vivo responses. However, impaired in vivo persistence of TCR-deficient CARs in mice might be different in humans, because the human cytokine mileu and lymphoid tissue are different.26,31 Given the diversity of human cytokine profiles in pretreated leukemia patients,32 and the differences in stromal cells and secondary lymphoid organs, mouse models hardly reflect completely the situation in patients. Despite these limitations of the animal model, differences of in vivo persistence and leukemia control both reflect T-cell functionality.
In conclusion, we present a novel and efficient tandem engineering approach to generate anti-CD19 CAR T cells with a CRISPR/Cas9-mediated KO of the TCRβ chain. TCR KO leads to significant reduction of alloreactivity both in vitro and in vivo. TCR KO in CAR T cells is associated with a reduction of in vivo persistence. Whereas allogeneic TCR−/CAR+ T cells show efficient early responses against leukemia cells, they might serve as temporary therapy until a successful generation of autologous CARs or bridge to transplant. The role and specificity of the endogenous TCR for the in vivo expansion of CAR T cells remains to be investigated in clinical studies and opens new options to improve longevity of remissions after CAR–T-cell therapy in the future.
For original data, please e-mail the corresponding author, Tobias Feuchtinger (tobias.feuchtinger@med.uni-muenchen.de).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Tanja Weißer, Nicola Habjan, Nadine Stoll, and Stefanie Stein for excellent technical assistance and Maike Fritschle, Annette Frank, and Liliana Mura for mouse handling.
This work was supported by the Kinderkrebshilfe Ebersberg e.V., Bettina Bräu Stiftung, Gesellschaft für Kinderkrebsforschung, Dr. Sepp und Hanne Sturm Gedaechtnisstiftung, Gertrud und Hugo Adler Stiftung, and Gottfried Kieser-Stiftung. S.W. was supported by the Else-Kröner-Fresenius Stiftung, and D.S. was supported by the German Cancer Research Center/German Cancer Consortium. S.K. is supported by the European Research Council (Starting Grant 756017). I.J. is funded by ERC Consolidator Grant 681524 and a Mildred Scheel Professorship from German Cancer Aid. The laboratory of T.G.P.G. is supported by grants from the Bettina Bräu Stiftung, the German Cancer Aid (DKH-70112257), and the Gert and Susanna Mayer Foundation.
Authorship
Contribution: D.S., T.A.S., T.K., S.W., and F.B. performed the experiments; D.S., F.B., and T.F. were responsible for concept development, analyzed the data, and prepared the manuscript; F.R., S.K., D.S., T.F., and F.B. designed CAR constructs and performed retroviral transduction; K.S., D.H.B., and T.A.S. designed gRNAs and established the CRISPR/Cas9 protocol; B.V. and I.J. performed in vivo experiments and established luciferase-transgenic PDX mouse models; B.W. performed irradiation; T.G.P.G. performed histology and immunohistochemistry; R.L. provided human serum; the visual abstract was created with BioRender.com; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: D.H.B. is cofounder of STAGE cell therapeutics GmbH (now Juno Therapeutics/BMS) and T cell factory B.V. (now Kite/Gilead); and has a consulting contract with and receives sponsored research support from Juno Therapeutics GmbH. The remaining authors declare no competing financial interests.
Correspondence: Tobias Feuchtinger, Department of Pediatric Hematology, Oncology, Hemostaseology, and Stem Cell Transplantation, Dr. von Hauner University Children’s Hospital, Ludwig Maximilian University Munich, Lindwurmstr 4, 80337 Munich, Germany; e-mail: tobias.feuchtinger@med.uni-muenchen.de.
