


Abstract
Glucose 6-phosphate dehydrogenase (G6PD) deficiency is 1 of the commonest human enzymopathies, caused by inherited mutations of the X-linked gene G6PD. G6PD deficiency makes red cells highly vulnerable to oxidative damage, and therefore susceptible to hemolysis. Over 200 G6PD mutations are known: approximately one-half are polymorphic and therefore common in various populations. Some 500 million persons with any of these mutations are mostly asymptomatic throughout their lifetime; however, any of them may develop acute and sometimes very severe hemolytic anemia when triggered by ingestion of fava beans, by any of a number of drugs (for example, primaquine, rasburicase), or, more rarely, by infection. Approximately one-half of the G6PD mutations are instead sporadic: rare patients with these mutations present with chronic nonspherocytic hemolytic anemia. Almost all G6PD mutations are missense mutations, causing amino acid replacements that entail deficiency of G6PD enzyme activity: they compromise the stability of the protein, the catalytic activity is decreased, or a combination of both mechanisms occurs. Thus, genotype-phenotype correlations have been reasonably well clarified in many cases. G6PD deficiency correlates remarkably, in its geographic distribution, with past/present malaria endemicity: indeed, it is a unique example of an X-linked human polymorphism balanced through protection of heterozygotes from malaria mortality. Acute hemolytic anemia can be managed effectively provided it is promptly diagnosed. Reliable diagnostic procedures are available, with point-of-care tests becoming increasingly important where primaquine and its recently introduced analog tafenoquine are required for the elimination of malaria.
G6PD deficiency
Among inborn errors of metabolism, the phrase by which inherited disorders were formerly known, glucose-6-phosphate dehydrogenase (G6PD) deficiency was the first red cell enzymopathy to be identified (Box 1), and it proved unique in at least 2 ways. First, G6PD deficiency is a polymorphic genetic trait with worldwide distribution (a current estimate is that over 500 million people are affected), but with great variation in prevalence, from 0 in the original Amerindian populations to 20% or more in parts of Africa and of Asia (Figure 1). Second, G6PD deficiency is a paradigmatic example of pharmacogenetics23 : nearly all people with this trait have no disease, indeed no signs or symptoms, unless and until they are exposed to an exogenous agent that triggers acute hemolytic anemia (AHA), which may be severe and even life-threatening.
G6PD and G6PD deficiency: historical milestones
1932. Otto Warburg and Walter Christian in Berlin, Germany, identified in yeast and in red cells an enzyme of carbohydrate metabolism that oxidized glucose-6-phosphate.1 Because the oxidation was not carried out by O2 itself, but required as an intermediary the coenzyme NADP (then called TPN), they named the enzyme Zwischenferment: we now know it was G6PD.
1956. Paul Carson in Alf Alving’s laboratory in Chicago, IL, discovered that men who had developed AHA following administration of the antimalarial primaquine had severe deficiency of G6PD in their red cells.2
1957. Gennaro Sansone and Giuseppe Segni in Genoa, Italy, found G6PD deficiency in all patients who had a history of favism.3
1958. Arieh Szeinberg and colleagues in Tel Aviv, Israel, found that the inheritance of G6PD deficiency was consistent with the gene being on the X chromosome.4
1961. Franco Panizon in Sassari, Italy,5 and Spyros Doxiadis in Athens, Greece,6 identified G6PD deficiency as a cause of severe neonatal jaundice.
1962. Shortly after the formulation of the “Lyon hypothesis” regarding inactivation of 1 of the 2 X chromosomes in somatic cells of female mice, Ernest Beutler at City of Hope, Duarte, CA, demonstrated that this created mosaicism in human blood cells in heterozygotes for G6PD deficiency.7
1965. Lucio Luzzatto and Normal Allan in Ibadan, Nigeria, found that G6PD A− was structurally different from G6PD A, rather than being a lower amount of the same.8
1966. The World Health Organization in Geneva, Switzerland, held an international Study Group in the aim to standardize methodologies for investigating G6PD deficiency and its genetic polymorphism.9
1967. Frank Livingstone in Michigan compiled in a book hundreds of data on the frequency of G6PD deficiency in human populations.10
1969. Philip Cohen and Michael Rosemeyer11 show that the active form of human G6PD is either a dimer or a tetramer of identical subunits.
1980. Barbara Migeon in Baltimore, MD, maps the G6PD gene to the tip of the long arm of the X chromosome (band Xq28), near the hemophilia A gene.12
1981. Maria Grazia Persico in Naples, Italy, clones a G6PD-related cDNA13 ; by 1986, through a collaboration between Naples and London, we had the entire cDNA sequence and the complete structure of the G6PD genomic gene.14
1995-2002. Embryonic stem cells in which G6PD had been knocked out by targeted homologous recombination have normal pentose synthesis but exquisite sensitivity to oxidative stress15 ; and G6PD inactivation is lethal early in embryo development.16
1999. Through work pioneered by Margaret Adams in Oxford, United Kingdom,17 with coworkers in London18 and in Hong Kong (SWN Au and Veronica Lam), the 3D structure of tetrameric human G6PD was solved by X-ray crystallography.19 Veronica Lam sadly died in the tsunami of 2004.
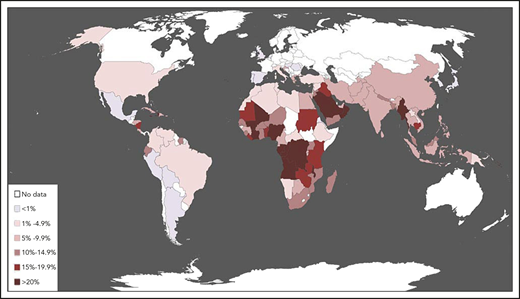
Epidemiology of G6PD deficiency. The map was constructed by compiling, from the world literature, data on the population frequency of polymorphic G6PD alleles that cause enzyme deficiency. Country-specific frequency ranges, full references, and additional details are given in supplemental Table 1, available on the Blood Web site. Although frequencies may vary widely within the same country, each country was assigned here a color based on the best estimate of the average frequency of G6PD deficiency, probably the most important figure from the point of view of public health. This map represents a thorough update of one first published in 1989 and widely reproduced.20 A similar map was published in 2009,21 and one based on a sophisticated geostatistical model in 2012.22
Epidemiology of G6PD deficiency. The map was constructed by compiling, from the world literature, data on the population frequency of polymorphic G6PD alleles that cause enzyme deficiency. Country-specific frequency ranges, full references, and additional details are given in supplemental Table 1, available on the Blood Web site. Although frequencies may vary widely within the same country, each country was assigned here a color based on the best estimate of the average frequency of G6PD deficiency, probably the most important figure from the point of view of public health. This map represents a thorough update of one first published in 1989 and widely reproduced.20 A similar map was published in 2009,21 and one based on a sophisticated geostatistical model in 2012.22
Biochemistry of G6PD and G6PD deficiency
G6PD is an enzyme very ancient in evolution, found in all organisms except for Archaea, which are mostly anaerobic, and a few obligate surface parasites (for example, Mycoplasma genitalium) and intracellular parasites (for example, Rickettsia prowazekii).24 G6PD is an oxidoreductase that catalyzes the oxidation of glucose-6-phosphate to 6-phosphoglucono-lactone coupled to the reduction of NAD phosphate (NADP) to reduced NADP (NADPH) (Figure 2). In all animals, G6PD is ubiquitously expressed, suggesting that it has an essential housekeeping function: indeed, knockout of the G6PD gene is lethal early in embryonic life.16 G6PD is often referred to as the first enzyme of the pentose phosphate pathway,28 underscoring its role in the production of pentose sugars required for nucleic acid synthesis (but pentose can also be produced through the alternative transketolase-transaldolase pathway). NADPH, produced by G6PD and called its coenzyme, is the electron donor in reactions required for the biosynthesis of deoxyribonucleotides, fatty acids, and steroids; it is also the coenzyme of cytochrome P450, central to the metabolism of many drugs and other xenobiotics. The reducing power of NADPH is required in what is commonly referred to as defense against oxidative challenges.28
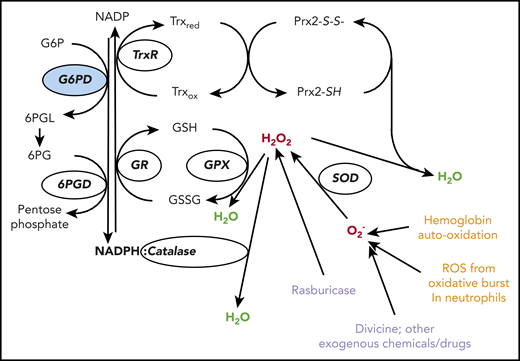
The red cell has several lines of defense against oxidative stress. Hydrogen peroxide (H2O2: in red) at the center, is a powerful oxidizing agent arising from superoxide (O2−) via superoxide dismutase (SOD). O2− (representing in this figure, in red, ROS in general) is a by-product of auto-oxidation of Hb within the red cells, and also of the oxidative burst in neutrophils (endogenous sources, in orange). In red cells, H2O2 can be detoxified to H2O (in green) by 3 different enzyme-mediated mechanisms. (i) Catalase directly degrades H2O2 to H2O: it has 2 to 4 molecules of NADPH in its structure.25 (ii) Glutathione peroxidase (GPX) catalyzes the same reaction coupled to the oxidation of reduced glutathione (GSH): it relies on the NADPH-linked glutathione reductase (GR) for regeneration of GSH. (iii) Peroxiredoxin-2 (Prx2) also degrades H2O2 at the expense of its own sulhydryl groups that become disulphides, and can be regenerated by thioredoxin (Trx) through thioredoxin reductase (TrxR).26 We do not know in quantitative terms the relative contributions of these 3 mechanisms to the degradation of H2O2 in human red cells under different conditions. However, we do know that acatalasemia is not associated with hemolytic anemia, whereas GR deficiency is associated with favism,27 supporting the importance of mechanism (ii); Prx2 deficiency in humans is not known, but it does cause hemolytic anemia in mice. Most important, all 3 mechanisms depend on NADPH, a steady supply of which can be provided in red cells only by G6PD (and 6PGD, which, however, depends on G6PD for supply of its own substrate; therefore, the crucial role of G6PD is highlighted in blue). Exogenous agents (in purple) can impose severe oxidative challenge by producing ROS or H2O2 directly; divicine, the aglycone of vicine (together with isouramil), is the chemical responsible for favism.
The red cell has several lines of defense against oxidative stress. Hydrogen peroxide (H2O2: in red) at the center, is a powerful oxidizing agent arising from superoxide (O2−) via superoxide dismutase (SOD). O2− (representing in this figure, in red, ROS in general) is a by-product of auto-oxidation of Hb within the red cells, and also of the oxidative burst in neutrophils (endogenous sources, in orange). In red cells, H2O2 can be detoxified to H2O (in green) by 3 different enzyme-mediated mechanisms. (i) Catalase directly degrades H2O2 to H2O: it has 2 to 4 molecules of NADPH in its structure.25 (ii) Glutathione peroxidase (GPX) catalyzes the same reaction coupled to the oxidation of reduced glutathione (GSH): it relies on the NADPH-linked glutathione reductase (GR) for regeneration of GSH. (iii) Peroxiredoxin-2 (Prx2) also degrades H2O2 at the expense of its own sulhydryl groups that become disulphides, and can be regenerated by thioredoxin (Trx) through thioredoxin reductase (TrxR).26 We do not know in quantitative terms the relative contributions of these 3 mechanisms to the degradation of H2O2 in human red cells under different conditions. However, we do know that acatalasemia is not associated with hemolytic anemia, whereas GR deficiency is associated with favism,27 supporting the importance of mechanism (ii); Prx2 deficiency in humans is not known, but it does cause hemolytic anemia in mice. Most important, all 3 mechanisms depend on NADPH, a steady supply of which can be provided in red cells only by G6PD (and 6PGD, which, however, depends on G6PD for supply of its own substrate; therefore, the crucial role of G6PD is highlighted in blue). Exogenous agents (in purple) can impose severe oxidative challenge by producing ROS or H2O2 directly; divicine, the aglycone of vicine (together with isouramil), is the chemical responsible for favism.
There is no nucleus and therefore no DNA or RNA synthesis, no fatty acid synthesis, no endoplasmic reticulum, and therefore no cytochrome P45029 in mature red cells: therefore, in their physiology, all of these factors are irrelevant except for the last function, which is paramount. Indeed, red cells are constantly exposed to an endogenous oxidative challenge because the concentration of hemoglobin (Hb) in red cells is no less than 5 mM. The reversible binding of O2 to the 4 heme residues within Hb is a masterpiece of physical chemistry, but no physical device can be perfect. Spontaneous conformational fluctuations in the heme pocket of HbO2 occasionally allow water or a small anion to enter, resulting in transfer of an electron from the iron to oxygen to produce methemoglobin and superoxide radicals: this auto-oxidation process affects 2% to 3% of total Hb each day (Low et al30 ). Methemoglobin (Fe3+) is rapidly reduced back to Hb (Fe2+) by NADH cytochrome b5 reductase 3 (also known as methemoglobin reductase); whereas superoxide radicals, via superoxide dismutase, produce hydrogen peroxide (H2O2), a powerful oxidizing agent. It is here that the provision of NADPH is critical: indeed, 3 enzymatic processes in the red cell can remove H2O2, and NADPH is involved in all 3 (Figure 2). In red cells, G6PD is the 1 key to facing oxidative challenge.
Because mature red cells have no protein synthesis, G6PD activity physiologically decreases as red cells age.31 Reticulocytes have a high level of G6PD activity, whereas only approximately one-tenth of that remains in the oldest red cells; however, in G6PD normal red cells, this amount is still sufficient for their needs.
AHA
Clinical manifestations
The commonest trigger of hemolysis in G6PD-deficient persons is a meal of fava beans: therefore, we briefly describe here an attack of favism (Figure 3A), a topic recently reviewed elsewhere.32 The patient is usually a boy in the 2- to 10-year age group (but girls and adults are not exempt as discussed in “Genetics and molecular genetics”) who appears pale, jaundiced, and quite ill with abdominal pain and sometimes fever. On examination, the spleen may be enlarged, and the urine is usually dark (“like red wine” or “like Coca-Cola,” depending on cultural preferences). Blood examination shows moderate to very severe anemia, and a rather spectacular blood smear (Figure 3B). Supravital staining with methyl violet reveals Heinz bodies; there is often a neutrophil leukocytosis with shift to the left; the platelets are usually normal. The unconjugated bilirubin and lactate dehydrogrenase are elevated; haptoglobin is low or undetectable.
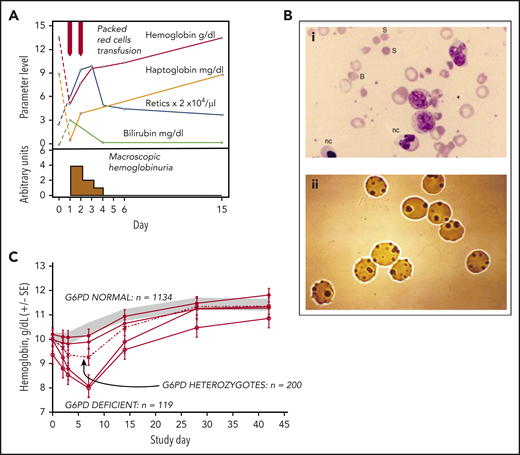
Clinical course of AHA in G6PD-deficient persons. (A) Attack of severe favism in a 5-year-old Sardinian boy (courtesy of Gianfranco Meloni). (B) Blood smears on day 1 of patients with favism: (i) May-Grünwald-Giemsa staining, original magnification ×1000; (ii) Heinz body supravital staining, original magnification ×1000. (C) Time course of Hb levels in a large cohort of children with P falciparum malaria who received antimalarial treatment with a drug combination (Lapdap) containing dapsone. Eleven percent of G6PD-deficient hemizygous boys and 0.5% of heterozygous girls required blood transfusion. All of these children were in clinical trials under appropriate medical supervision; there were no deaths (there might have been outside of clinical trials). B, bite cell; nc, nucleated red cell; S, spherocyte. Modified from (A) Luzzatto and Arese,32 (B) Luzzatto and Poggi,63 and (C) Pamba et al33 with permission.
Clinical course of AHA in G6PD-deficient persons. (A) Attack of severe favism in a 5-year-old Sardinian boy (courtesy of Gianfranco Meloni). (B) Blood smears on day 1 of patients with favism: (i) May-Grünwald-Giemsa staining, original magnification ×1000; (ii) Heinz body supravital staining, original magnification ×1000. (C) Time course of Hb levels in a large cohort of children with P falciparum malaria who received antimalarial treatment with a drug combination (Lapdap) containing dapsone. Eleven percent of G6PD-deficient hemizygous boys and 0.5% of heterozygous girls required blood transfusion. All of these children were in clinical trials under appropriate medical supervision; there were no deaths (there might have been outside of clinical trials). B, bite cell; nc, nucleated red cell; S, spherocyte. Modified from (A) Luzzatto and Arese,32 (B) Luzzatto and Poggi,63 and (C) Pamba et al33 with permission.
When a drug is the trigger (Table 1), the clinical picture is similar. The largest set of patients on whom there is full documentation is that of children treated with an antimalarial combination that contained dapsone (Figure 3C). Like in favism, hemolysis was brisk, with the hemoglobin nadir on day 333 ; but, unlike in favism, all children were already anemic (from malaria and/or other causes) when they were exposed to dapsone: this illustrates the importance of comorbidity in the clinical assessment of AHA. Drug-induced AHA is, by definition, iatrogenic: therefore, in preventing it, we bear an extra responsibility, which has become particularly poignant with the introduction of rasburicase and its long-acting derivative pegloticase.34 These drugs are used electively in patients at risk for tumor lysis syndrome or kidney injury: they are contraindicated in patients with G6PD deficiency, meaning that there is no justification for using them before testing for G6PD deficiency (Box 2). There is a tendency to limit testing to patients from ethnic groups in which G6PD deficiency is common. Given that ethnic group is a vague concept and that ancestry may be mixed or only partly known, we recommend that, before giving rasburicase, G6PD testing should always be done.
Development of diagnostic tests for G6PD deficiency
Since the discovery of G6PD deficiency the standard spectrophotometric assay of G6PD enzyme activity (Box 1), used on hemolysates, has provided a highly reliable diagnostic test.40
Brewer41 observed that after Hb (Fe2+) in intact red cells is converted to methemoglobin (Fe3+) by the addition of sodium nitrite, reversal to Hb (Fe2+) depends on the G6PD activity within the red cells themselves: G6PD normal samples will return to bright red, whereas G6PD-deficient samples will stay brown.
Motulsky and Campbell-Kraut42 observed that the time for brilliant cresyl blue (BCB) added to hemolysates to become colorless was roughly proportional to G6PD activity.
Both the BCB and the methemoglobin reduction tests lent themselves to processing large numbers of samples: they were the first G6PD screening tests, and they were widely used, for instance, to determine the frequency of G6PD deficiency in various human populations (Livingstone10 ).
Based on the screening method carried out on intact red cells (no. 2 in this list), a cytochemical test was developed in 1965.43 Under certain precise conditions methemoglobin is eluted from red cells but Hb is not: thus, after smearing red cells on a slide, elution and staining, G6PD deficiency can be quantitatively assessed by a differential count of G6PD normal and G6PD-deficient red cells.
From 1966 to 1968, Beutler’s laboratory developed a “spot test” for the diagnosis of G6PD deficiency that could be carried out by just applying to a piece of filter paper 1 reagent mixture and 10 μL of blood: after 15 minutes the spot will show bright fluorescence only if the sample is G6PD normal.44 This fluorescent spot test (FST) became popular not only for work in the field but also in diagnostic laboratories because it is reliable, inexpensive, and easy (the only equipment required is a UV lamp).
A new cytochemical test, based on the use of tetrazolium dyes, was introduced in 1968 by Fairbanks and Lampe45 in 1968; subsequently the test was perfected by Van Noorden.46,47
Both cytochemical methods (nos. 5 and 7 in this list) have been used particularly in testing women heterozygous for G6PD deficiency, who have therefore a mosaic composition of blood cells; the method mentioned under item 7 has proven to be technically easier.
Although screening methods were already available [nos. 2, 3, and 6 in this list], the resurgence of primaquine and the introduction of tafenoquine,48 for the administration of which G6PD testing is label-mandatory, have been a strong stimulus to the development of point of care (POC) tests. These are becoming available as commercial kits: some are suitable for identifying not only G6PD-deficient hemizygotes and homozygotes, but also heterozygous females at risk of AHA.49
Given that many G6PD mutations are known (Table 2; Figure 5), a G6PD-deficiency status can be inferred reliably by mutation analysis (genotypic testing). This approach has the advantage that it can be carried out on stored DNA, and it circumvents the risk of misclassification that arises when reticulocytosis produces in a G6PD-deficient person an increase in G6PD enzyme activity that may, though rarely, be so marked as to reach the normal range. However, there are 2 caveats: (a) one needs to know which mutations are prevalent in the population to which the patient belongs; and one may still miss an unusual or unknown mutation; (b) in heterozygous females, on account of the wide range of the ratio between G6PD normal and G6PD-deficient red cells generated by X-inactivation, the genotype does not predict the degree of G6PD deficiency: from this point of view, in heterozygotes, a phenotypic test is superior to a genotypic test.
Drugs that may trigger AHA in G6PD-deficient patients
| Definite risk of AHA | “Possible” risk of AHA | Possible association (less likely) |
|---|---|---|
| Antimalarial drugs | ||
| Dapsone-containing combinations* | Chloroquine | |
| Pamaquine* | Quinidine | |
| Primaquine* | Quinine | |
| Tafenoquine | ||
| Other drugs | ||
| Ciprofloxacin | Aspirin† | Chloramphenicol |
| Glibenclamide‡ | Menadiol sodium phosphate | Dimercaptosuccinic acid |
| Methylthioninium chloride*,§ | Sulfadiazine | Glibenclamide |
| Moxifloxacin | Sulfasalazine | Mepacrine |
| Nalidixic acid | Sulfonylureas | Vitamin K analogs |
| Niridazole | Tolonium chloride|| | |
| Nitrofurantoin*,¶ | ||
| Norfloxacin | ||
| Ofloxacin | ||
| Phenazopyridine# | ||
| Rasburicase and pegloticase34,* | ||
| Sulfamethoxazole/ cotrimoxazole | ||
| Henna (cosmetic use)35-37 |
| Definite risk of AHA | “Possible” risk of AHA | Possible association (less likely) |
|---|---|---|
| Antimalarial drugs | ||
| Dapsone-containing combinations* | Chloroquine | |
| Pamaquine* | Quinidine | |
| Primaquine* | Quinine | |
| Tafenoquine | ||
| Other drugs | ||
| Ciprofloxacin | Aspirin† | Chloramphenicol |
| Glibenclamide‡ | Menadiol sodium phosphate | Dimercaptosuccinic acid |
| Methylthioninium chloride*,§ | Sulfadiazine | Glibenclamide |
| Moxifloxacin | Sulfasalazine | Mepacrine |
| Nalidixic acid | Sulfonylureas | Vitamin K analogs |
| Niridazole | Tolonium chloride|| | |
| Nitrofurantoin*,¶ | ||
| Norfloxacin | ||
| Ofloxacin | ||
| Phenazopyridine# | ||
| Rasburicase and pegloticase34,* | ||
| Sulfamethoxazole/ cotrimoxazole | ||
| Henna (cosmetic use)35-37 |
Modified from Table 4 of Luzzatto et al,38 where sources and additional references are given.
Evidence-based according to Youngster et al.39
Acetylsalicylic acid. Like for all drugs, hemolysis is dose-related. The regimen of 75 to 100 mg per day, widely used for the prophylaxis of cardiovascular events, is safe for G6PD-deficient persons.
Glyburide.
Methylene blue.
Toluidine blue.
Furadantin.
Pyridium.
Infection is the third potential trigger of AHA, but it is more erratic. Broadly speaking, bacterial infection must be severe to cause AHA: it has been reported with pneumonia,50 brucellosis,51 rickettsiosis,52 maxillary abscesses caused by Streptococcus,53 or Staphylococcus,54 and even with Clostridium difficile55 infection. AHA also occurs in the course of hepatitis,56 including hepatitis A, B,57 and E.58,59 With viral hepatitis in G6PD-deficient patients, hyperbilirubinemia is sometimes excessive even when there is little or no evidence of AHA,56-60 and acute renal failure can be a further complication.60 AHA has also been reported occasionally in the course of cytomegalovirus infection61 and in 1 instance of dengue fever.62 AHA triggered by infection can be quite severe: if it subsides when the infection is controlled, then we can be confident that infection was the trigger. In some cases, AHA has been mistakenly blamed, rather than on the infection, on an antibiotic used to treat it; but the reverse could also happen.
With both fava beans and drugs, G6PD deficiency-related hemolysis is characteristically dose-dependent: the cases that come to the emergency room are the “tip of the iceberg.” When the anemia is mild, the patient may not be seen in hospital and may often be undiagnosed; in many cases, there may not even be anemia, but only compensated hemolysis.
Diagnosis and management
If AHA is severe, blood transfusion may be urgent63 ; otherwise, only fluid support and analgesics may be needed. In most cases, the patient had been previously asymptomatic: AHA will prompt testing for G6PD deficiency, which should be done by a quantitative test (Box 2). In most cases, the result will be clear cut, that is, below the normal range of 7 to 11 IU/g Hb.64 Reference ranges vary, and unfortunately those stated by some laboratories are too wide. A result below 80% of the lower limit of normal must be regarded as G6PD deficient. Sometimes, during or immediately after a hemolytic attack, G6PD activity from a G6PD-deficient patient may be within the “normal range” (as a result of both destruction of the oldest cells and reticulocytosis). In such cases, the test must be repeated after a few weeks, or a known mutation can be revealed by DNA testing.
In areas with high prevalence, donor blood may be G6PD deficient: in general, this can be used safely65 and universal screening of donors is not practical. Ideally, it would of course be preferable to use G6PD normal blood when needed for AHA in a G6PD-deficient patient, when doing an exchange transfusion for neonatal jaundice (NNJ; see “NNJ” later in text), or in patients requiring regular blood transfusion.
Clinical course
Unlike with autoimmune AHA, which often poses considerable therapeutic challenges, with G6PD-related AHA, rapid recovery is the rule. A most striking feature is the speed of the reticulocyte response (Figure 3A). It is hard to imagine that a hypoxia-erythropoietin–mediated response can take place within 48 hours: there must be a rush order (mechanism not yet known) that prompts the exiting from the marrow of existing reticulocytes.
Pathophysiology
This type of AHA seems almost like experimental hematology (reminiscent of a phenylhydrazine-treated rabbit).66 The zero time is exposure to the trigger; from then on, we witness what happens when, in a person with a previously normal blood count, up to two-thirds of red cells are suddenly destroyed by a process referred to as oxidative damage. The culprit is a chemical (primaquine, or any of the other drugs listed in Table 1, or divicine and isouramil from fava beans) that can generate reactive oxygen radicals (ROS) and H2O2 (Figure 2): these can also be produced directly in the course of bacterial infection. As noted in the previous section, ROS are generated all the time through auto-oxidation of Hb; however, the rate at which they are thus produced is low, so that even G6PD-deficient red cells can provide enough NADPH to cope with them. When the levels of ROS are instead higher, red cells with normal G6PD respond by recycling NADPH at an increased rate, thereby preventing damage. G6PD-deficient red cells are unable to do so, which results in formation of ferryl hemoglobin and of hemichromes (partially denatured hemoglobin); at the same time, lipids and thiol groups in cytoplasmic and membrane proteins are oxidized. Binding of hemichromes to the membrane cytoskeleton leads to formation of Heinz bodies; aggregation of membrane proteins leads to formation of cross-bonded rigid hemighosts.32,67 Glutathione (GSH) plays a special role in defense against oxidative damage because, in addition to being the substrate of GSH peroxidase, it can also regenerate thiol groups in proteins that had been oxidized to disulphides.68 The more severely damaged red cells will undergo intravascular hemolysis, with consequent hemoglobinuria. In other red cells, clusters of oxidized band 3 in the membrane will bind immunoglobulin G and complement factor C3c69 ; thus, opsonized, they will undergo erythrophagocytosis, that is, extravascular hemolysis. Intravascular hemolysis accounts, of course, for hemoglobinuria, and also for abdominal pain, because plasma Hb will bind nitric oxide causing smooth muscle dystonia; extravascular hemolysis accounts for jaundice and for splenomegaly. In G6PD-deficient red cells, like in normal red cells, there is a downward gradient in G6PD activity as they age70 : therefore, the oldest red cells will be the first to suffer damage; whereas the youngest red cells, including reticulocytes, are relatively resistant to hemolysis.
Chronic nonspherocytic hemolytic anemia
This rather cumbersome heading originates from history. Hereditary spherocytosis (HS) has been, for over a century, a prototype of congenital hemolytic anemias (other than hemoglobinopathies), but in some patients with this kind of condition, spherocytes may not be prominent on a blood smear: after G6PD deficiency was discovered, some of them were found to be G6PD deficient.
In contrast to the high prevalence of G6PD deficiency that entails the risk of AHA, chronic nonspherocytic hemolytic anemia (CNSHA) is a rare disease (estimated frequency, <10 per million). The clinical picture is generally similar to that of HS, including jaundice and gallstones, with wide-ranging severity (see examples in Table 2): from mild (for example, diagnosed incidentally in an adult) to severe enough (in a minority of patients) to require recurrent blood transfusion. Unlike in HS, a history of NNJ (see next section), often severe, is the rule. Needless to say, all agents capable of causing AHA in any G6PD-deficient person will cause acute on chronic hemolysis in patients with CNSHA. In some transfusion-dependent patients, splenectomy has proven highly beneficial (Figure 4).
Different G6PD mutations produce a wide range of biochemical phenotypes
| G6PD variant | Enzyme activity in red cells, % of normal | Electro phoretic mobility | Thermal stability in vitro* | KmG6P, μM | kcat s−1 | Amino acid change | Class | Clinical features | Reference/notes |
|---|---|---|---|---|---|---|---|---|---|
| B (“wild type”) | 100 | Normal | Normal | 50-70 | 275 | — | IV | None | 71 |
| A | 85 | Fast | Normal | 60 | 247 | N126D | IV | None | 72 |
| A− | 10-23 | Fast | Low | 47-60 | 137 |
| III | AHA | 8: Thermostability low at low [NADP]; 73: impaired folding observed |
| Orissa | 13-28 | Normal | Increased | 135 | A44G | III | AHA | 74 | |
| Seattle | 17 | Slow | Normal | 20 | 207† | D282H | III | AHA | 70: Accelerated decay in vivo |
| Mediterranean | <5 | Normal | Low | 12-23 | 98† | S188F | II | AHA | 70: Accelerated decay in vivo |
| Andalus | 1 | Low | 14 | 40 | R454H | II | AHA | 71 | |
| Union | 3 | Fast (exp) | Low | 10 | 29 | R454C | II | AHA | 71 |
| Canton | 4 | Fast (103%) | Slightly decreased | 28-23 | 123 | R459L | II | AHA | 75; 76: first 3D structure of a human G6PD variant |
| Chatham | <2 | Normal | Low | 60 | A335T | II | AHA | 77 | |
| Viangchan | 3 | Normal | Normal | 42 | 145 | V291M | II | AHA | 78 |
| Mahidol | 0.6 | Normal | Normal | 75 | 207 | G163S | II | AHA | 79 |
| Plymouth | <1‡ | Fast (exp) | Slightly decreased | 67 | 249 | G163D | I | CNSHA, mild | 79,80: Also cardiomyopathy |
| Nilgiri | 2 | Low | 81 | 3 | R198H | II | AHA | 81 | |
| Santiago | <1 | Fast (exp) | Very low | 26 | 4 | R198P | I | CNSHA | 82 |
| Nashville Portici | 1 | Slow | Very low | 144 | 119 | R393H | I | Severe NNJ; CNSHA, moderate; hepatosplenomegaly; BT+ | 83: Accelerated decay in vivo |
| Campinas | <1‡ | Normal (exp) | Very low | 81 | 212 | G488V | I | Severe NNJ; CNSHA, severe; BT-dependent | 84 |
| Harilaou | <1‡ | Normal | Very low | 90 | F216L | I | Severe NNJ; CNSHA, severe, BT-dependent until splenectomized | 85: Stabilized in interspecific hybrid molecule | |
| Volendam | 1 | Slow | Low | 238 | P172S | I | NNJ; mild anemia with infection-related exacerbations | 86: Heterozygous woman with skewed X-inactivation with de novo mutation |
| G6PD variant | Enzyme activity in red cells, % of normal | Electro phoretic mobility | Thermal stability in vitro* | KmG6P, μM | kcat s−1 | Amino acid change | Class | Clinical features | Reference/notes |
|---|---|---|---|---|---|---|---|---|---|
| B (“wild type”) | 100 | Normal | Normal | 50-70 | 275 | — | IV | None | 71 |
| A | 85 | Fast | Normal | 60 | 247 | N126D | IV | None | 72 |
| A− | 10-23 | Fast | Low | 47-60 | 137 |
| III | AHA | 8: Thermostability low at low [NADP]; 73: impaired folding observed |
| Orissa | 13-28 | Normal | Increased | 135 | A44G | III | AHA | 74 | |
| Seattle | 17 | Slow | Normal | 20 | 207† | D282H | III | AHA | 70: Accelerated decay in vivo |
| Mediterranean | <5 | Normal | Low | 12-23 | 98† | S188F | II | AHA | 70: Accelerated decay in vivo |
| Andalus | 1 | Low | 14 | 40 | R454H | II | AHA | 71 | |
| Union | 3 | Fast (exp) | Low | 10 | 29 | R454C | II | AHA | 71 |
| Canton | 4 | Fast (103%) | Slightly decreased | 28-23 | 123 | R459L | II | AHA | 75; 76: first 3D structure of a human G6PD variant |
| Chatham | <2 | Normal | Low | 60 | A335T | II | AHA | 77 | |
| Viangchan | 3 | Normal | Normal | 42 | 145 | V291M | II | AHA | 78 |
| Mahidol | 0.6 | Normal | Normal | 75 | 207 | G163S | II | AHA | 79 |
| Plymouth | <1‡ | Fast (exp) | Slightly decreased | 67 | 249 | G163D | I | CNSHA, mild | 79,80: Also cardiomyopathy |
| Nilgiri | 2 | Low | 81 | 3 | R198H | II | AHA | 81 | |
| Santiago | <1 | Fast (exp) | Very low | 26 | 4 | R198P | I | CNSHA | 82 |
| Nashville Portici | 1 | Slow | Very low | 144 | 119 | R393H | I | Severe NNJ; CNSHA, moderate; hepatosplenomegaly; BT+ | 83: Accelerated decay in vivo |
| Campinas | <1‡ | Normal (exp) | Very low | 81 | 212 | G488V | I | Severe NNJ; CNSHA, severe; BT-dependent | 84 |
| Harilaou | <1‡ | Normal | Very low | 90 | F216L | I | Severe NNJ; CNSHA, severe, BT-dependent until splenectomized | 85: Stabilized in interspecific hybrid molecule | |
| Volendam | 1 | Slow | Low | 238 | P172S | I | NNJ; mild anemia with infection-related exacerbations | 86: Heterozygous woman with skewed X-inactivation with de novo mutation |
From the full list (supplemental Table 2) of G6PD variants we have selected, among those for which enzymatic properties are known more extensively, some polymorphic (classes II-IV) and some rare (class I) variants. These properties were reported either in Betke et al9 or when the variant was originally described (references in supplemental Table 2), or in subsequent papers (references in this table). Class I variants are, by definition, associated with CNSHA: but within this diagnosis there is a wide spectrum of severity.
—, no change; 3D, 3-dimensional; BT, blood transfusion; CNSHA, chronic nonspherocytic hemolytic anemia; exp, expected.
Thermostability studies must be carried out on purified or recombinant enzyme, and they have been carried out in different ways, making comparisons problematic: hence, we have adopted in this table a semiquantitative terminology. Rather than the time it takes to inactivate the enzyme at a fixed temperature, it is probably more informative to determine T1/2 (temperature at which 50% activity is lost after a fixed exposure time, eg, 7 minutes): this parameter was first introduced in 19658 and found to be highly sensitive to the concentration of NADP. The properties of this arbitrary set of variants illustrate the following: (1) Almost all class I variants show evidence of markedly impaired stability; several of the others are also unstable, and in some this has been found to correlate with accelerated decay in vivo; (2) KmG6P, when increased, affects performance in the steady state; therefore, it is not surprising to find this feature in some class I variants (for example, Portici, Volendam). G6PD Orissa is not in class I (despite high KmG6P) probably thanks to its relatively high residual activity; (3) To determine kcat, pure enzyme (usually obtained by recombinant DNA technology) is required (hence some data are missing): it is moderately decreased in many variants; drastically decreased in G6PD Nilgiri and G6PD Santiago; (4) There is overlap in residual enzyme activity values between class II and class I; however, as a rule, all class I variants have very low activity (1% or less: sometimes undetectable);(5) Comparing variants where the same amino acid is replaced: G6PD Plymouth (class I) differs from G6PD Mahidol (class II) only in reduced thermostability; interestingly, the amino acid replacement in G6PD Plymouth entails a change in charge, whereas that in G6PD Mahidol does not. G6PD Santiago (class I) and G6PD Nilgiri (class II) again differ only in thermostability; and again there is a charge change in the former but not in the latter.
Calculated from ratio of activity/cross reacting material compared with G6PD B.
Enzyme activity reported as undetectable.
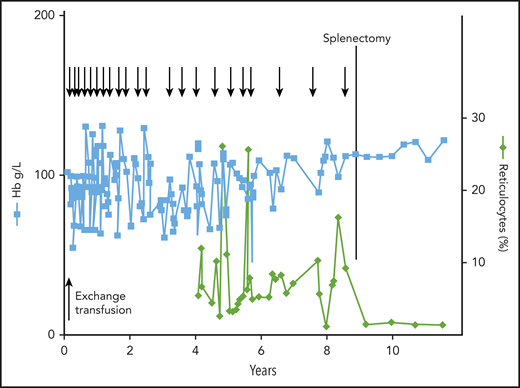
Clinical course of a patient with CNSHA. The patient presented with severe NNJ requiring exchange blood transfusion; he then had moderate to severe anemia requiring frequent blood tranfusions (arrows at the top). At 9 years of age, he was splenectomized, whereupon he became transfusion-independent. Note the persistent reticulocytosis, that remitted after splenectomy. The underlying unique variant was G6PD Harilaou (class I: F216L). Reproduced from Luzzatto and Poggi63 with permission.
Clinical course of a patient with CNSHA. The patient presented with severe NNJ requiring exchange blood transfusion; he then had moderate to severe anemia requiring frequent blood tranfusions (arrows at the top). At 9 years of age, he was splenectomized, whereupon he became transfusion-independent. Note the persistent reticulocytosis, that remitted after splenectomy. The underlying unique variant was G6PD Harilaou (class I: F216L). Reproduced from Luzzatto and Poggi63 with permission.
The clinical picture of CNSHA is so different from asymptomatic G6PD deficiency that it prompted early on9 a classification of underlying variants (Table 3). It is now clear that different mutations underlie these 2 phenotypes, as discussed later in text.
Molecular basis of G6PD variants
| Class I* | Classes II* and III* | Class IV* | Class not defined | Total | |
|---|---|---|---|---|---|
| Single aa replacement | 82† | 87 | 5‡ | 24 | 198 |
| 2 or 3 aa replacements | 6§ | 12|| | 18¶ | ||
| In-frame deletions | 11 | 11 | |||
| Other** | 2 | 1 | 3 | ||
| Total | 101† | 99 | 5 | 25 | 230 |
| Class I* | Classes II* and III* | Class IV* | Class not defined | Total | |
|---|---|---|---|---|---|
| Single aa replacement | 82† | 87 | 5‡ | 24 | 198 |
| 2 or 3 aa replacements | 6§ | 12|| | 18¶ | ||
| In-frame deletions | 11 | 11 | |||
| Other** | 2 | 1 | 3 | ||
| Total | 101† | 99 | 5 | 25 | 230 |
Details and references regarding all 230 G6PD protein variants with known mutation are given in supplemental Table 2.
According to a classification proposed in 197187 and universally used since, class IV variants have normal activity; class III variants have enzyme deficiency with residual activity 10% to 60%; class II variants have enzyme deficiency with residual activity <10%; class I variants are those that cause CNSHA. We prefer to consider class II and class III variants as 1 group38 because their clinical manifestations are the same: even though, when they occur, they tend to be more severe, on average and with wide overlap, with class II variants than with class III variants. In view of this, the WHO is currently considering a revision of this classification.
There is 1 example of 2 different mutations resulting in the same amino acid change (F173L); there is 1 example of 2 contiguous base replacements within the same codon (G6PD Palermo); in 2 variants, 2 mutations occur in the same codon; in 1 case, this is the only mutation; in another case, it is associated with another 2 mutations in the nearby codons (G6PD Crispim). Two variants (G6PD Tokyo Glu416Lys, G6PD Herlev Arg198Ser) are reported as class I/II.
Until recently, the only class IV variant was G6PD A (originally characterized because electrophoretically fast compared with the “wild-type” G6PD B). Interestingly, 4 additional class IV variants have been recently discovered in a normal subjects database81 ).
Two variants with 3-aa replacements
One variant with 3-aa replacements
Fifteen variants with 2 and 3 variants with 3-aa replacements. Only 8 of the variants with multiple amino acid replacements carry an amino acid replacement not found as a single amino acid replacement: these specific amino acid replacements are 11.
Two variants with splicing site mutation and 1 with a nonsense mutation.
NNJ
In newborns, G6PD deficiency entails the risk of NNJ (Box 1). The vast pertinent literature (see Luzzatto and Poggi63 and Kaplan and Hammerman88 ) cannot be fully reviewed here, but some facts must be noted. First, NNJ in G6PD-deficient babies can be severe enough to cause kernicterus, therefore, prompt management, which may require exchange blood transfusion, is imperative if permanent neurologic damage is to be avoided. Second, unlike with severe NNJ caused, for example, by Rhesus incompatibility, in most cases there is little if any evidence of hemolysis: indeed, we have to admit that the mechanism is not yet clear.89 Third, although not all G6PD-deficient babies develop NNJ, in numerous studies from different parts of the world (for example, in Africa), the frequency of NNJ has been always higher in G6PD-deficient babies compared with G6PD normal controls.90,91 The coexistence of a UGT1A1 promoter mutation further increases this frequency. Fourth, the peak incidence of hyperbilirubinemia is on day 2 to day 3 after birth: this is of great practical importance because mother and baby may have been discharged already, and therefore there may be a delay or even a failure of a diagnosis that requires urgent management by phototherapy or exchange blood transfusion.
Genetics and molecular genetics
The G6PD gene (which maps to Xq28) has 13 exons (exon 1 being noncoding: Figure 5), and it encodes a polypeptide chain of 514 amino acids, the dimer and the tetramer of which (Figure 6) are the enzymatically active forms of G6PD (the monomer has no enzyme activity).
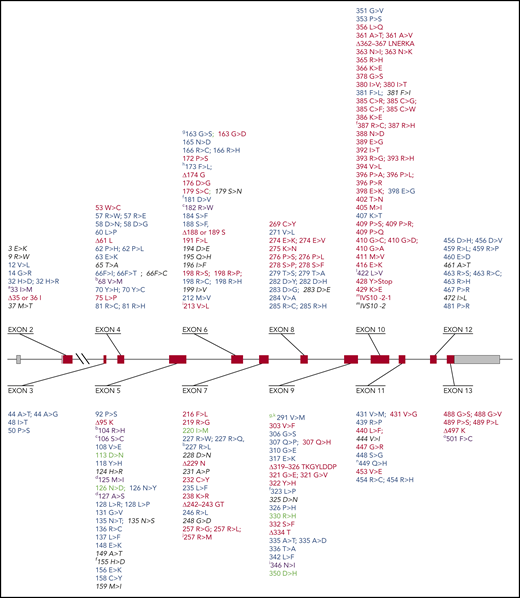
Known mutations in the G6PD gene. The full list of known mutations in the G6PD gene can be found in supplemental Table 2. The G6PD genomic gene spans ∼18 kb. Exons are shown as thick numbered blocks and introns as thin lines. Exon 1 (noncoding) and the 3′ untranslated region of exon 13 are in gray. The long intron 2 is indicated by a diagonal double line. Mutations with normal enzyme activity (class IV) are in green. Mutations that cause susceptibility to AHA (class II/III) are in blue. Mutations that cause CNSHA (class I) are in red. Mutations that have been observed only in association with another mutation are in purple. Mutations with unknown clinical features are in in black italics. The amino acid replacements caused by mutations are shown using the amino acid 1-letter symbols. Note the predominance of class I variants in exons 10 and 13. Additional notes: (a) This variant has, in addition to the mutation shown, also the mutation (454 Arg>Cys) of G6PD Union. (b) These variants have, in addition to the mutation shown, also the mutation (126 Asn>Asp) of G6PD A. (c) This variant has been reported to have 3 different mutations: 2 are unique (106 Ser>Lys and 182 Arg>Trp), whereas 1 is the mutation (198 Arg>Cys) of G6PD Coimbra. (d) This variant has been reported to have 3 different mutations: 2 are unique (125 Met>Ile and 127 Ala>Pro), whereas 1 is the mutation (128 Leu>Pro) of G6PD Vanua Lava. (f) This mutation has been found also associated with the mutation (126 Asn>Asp) of G6PD A. (g) These variants have, in addition to the mutation shown, also the mutation (291 Val>Met) of G6PD Viachang. (h) This variant is generated by 2 mutations within the same codon. (i) This variant has 2 unique mutations: 213 Val>Leu and 346 Asn>Ile. (j) This variant has 2 point mutations within the same codon. Δ indicates deletion. ∫, abnormal splicing; Ins, insertion.
Known mutations in the G6PD gene. The full list of known mutations in the G6PD gene can be found in supplemental Table 2. The G6PD genomic gene spans ∼18 kb. Exons are shown as thick numbered blocks and introns as thin lines. Exon 1 (noncoding) and the 3′ untranslated region of exon 13 are in gray. The long intron 2 is indicated by a diagonal double line. Mutations with normal enzyme activity (class IV) are in green. Mutations that cause susceptibility to AHA (class II/III) are in blue. Mutations that cause CNSHA (class I) are in red. Mutations that have been observed only in association with another mutation are in purple. Mutations with unknown clinical features are in in black italics. The amino acid replacements caused by mutations are shown using the amino acid 1-letter symbols. Note the predominance of class I variants in exons 10 and 13. Additional notes: (a) This variant has, in addition to the mutation shown, also the mutation (454 Arg>Cys) of G6PD Union. (b) These variants have, in addition to the mutation shown, also the mutation (126 Asn>Asp) of G6PD A. (c) This variant has been reported to have 3 different mutations: 2 are unique (106 Ser>Lys and 182 Arg>Trp), whereas 1 is the mutation (198 Arg>Cys) of G6PD Coimbra. (d) This variant has been reported to have 3 different mutations: 2 are unique (125 Met>Ile and 127 Ala>Pro), whereas 1 is the mutation (128 Leu>Pro) of G6PD Vanua Lava. (f) This mutation has been found also associated with the mutation (126 Asn>Asp) of G6PD A. (g) These variants have, in addition to the mutation shown, also the mutation (291 Val>Met) of G6PD Viachang. (h) This variant is generated by 2 mutations within the same codon. (i) This variant has 2 unique mutations: 213 Val>Leu and 346 Asn>Ile. (j) This variant has 2 point mutations within the same codon. Δ indicates deletion. ∫, abnormal splicing; Ins, insertion.
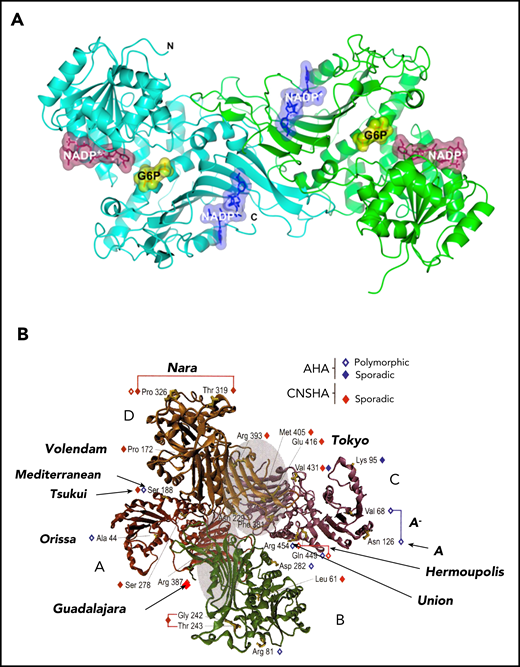
3-Dimensional structure of the human G6PD protein. Both the dimer and the tetramer are enzymatically active. The dimer/tetramer ratio within different cells or under different conditions is still not known, but it affects the pH-dependence of G6P binding.92 It is still not known what proportion of G6PD, within cells, is in dimer form. (A) The G6PD dimer, showing the binding sites of the 2 substrates G6P (yellow) and NADP (purple), and the site of the “structural” NADP (blue) (from Gómez-Manzo et al78 ). (B) The G6PD tetramer, showing the positions of a small selection of G6PD variants. Note (on top) the relatively large in-frame deletion of G6PD Nara: this is compatible with residual G6PD activity because the 8 aa involved are in a nonstructured region. Modified from Mason et al93 with permission.
3-Dimensional structure of the human G6PD protein. Both the dimer and the tetramer are enzymatically active. The dimer/tetramer ratio within different cells or under different conditions is still not known, but it affects the pH-dependence of G6P binding.92 It is still not known what proportion of G6PD, within cells, is in dimer form. (A) The G6PD dimer, showing the binding sites of the 2 substrates G6P (yellow) and NADP (purple), and the site of the “structural” NADP (blue) (from Gómez-Manzo et al78 ). (B) The G6PD tetramer, showing the positions of a small selection of G6PD variants. Note (on top) the relatively large in-frame deletion of G6PD Nara: this is compatible with residual G6PD activity because the 8 aa involved are in a nonstructured region. Modified from Mason et al93 with permission.
The consequences of X-linkage are very important. First, because a male has only 1 G6PD allele, he can only be either hemizygous G6PD normal or hemizygous G6PD deficient; a female instead, having 2 G6PD alleles, can be homozygous G6PD normal, or homozygous G6PD deficient, or heterozygous for G6PD deficiency. Second, in keeping with the laws of population genetics, in any given population, heterozygotes are more frequent than hemizygous G6PD-deficient males, whereas homozygous G6PD-deficient females are much more rare (see examples in Luzzatto et al38 ). Third, because G6PD, like most X-linked genes, is subject to the phenomenon of X-chromosome inactivation (Box 1), heterozygous females are epigenetic mosaics: in their blood, there is a mixture of G6PD normal and G6PD-deficient red cells. The ratio between the 2 types has a modal value of 1 (ie, 50% of each type), but this ratio is highly variable from 1 person to another: therefore, the phenotype of heterozygous females ranges from G6PD normal to G6PD, just as deficient as that of a hemizygous male. Some of these facts are sometimes misunderstood. At the population level, many studies report the frequency of G6PD deficiency in a sex-pooled population sample. This is unfortunate because only a subset of G6PD-deficient heterozygous females will have been classified as G6PD deficient, and therefore, in that population, we will not know the true frequency of the G6PD-deficient allele(s): instead, this could have been exactly ascertained from the frequency of G6PD-deficient males alone, which is identical to the frequency of the respective allele. It is also often stated that G6PD deficiency “is more frequent in males,” or “more expressed in males”: both statements are not correct. It is true, instead, that whereas G6PD deficiency is regularly expressed in G6PD-deficient hemizygous males and homozygous females, in heterozygous females, the expression, and therefore the potential severity of hemolysis, is highly variable. Surprisingly, G6PD deficiency is often qualified, even in textbooks, as “X-linked recessive”: of course this is wrong because G6PD deficiency is frequently expressed in heterozygotes, both biochemically and clinically.
There are now 230 G6PD variants with known mutations (Table 3; Figure 5; supplemental Table 2). All but 2 of them are either missense mutations (each 1 causing a single amino acid replacement) or small in-frame deletions (causing loss of 1 or a few amino acids): these will produce a qualitatively abnormal G6PD protein, which still has some residual enzyme activity. Frame-shift mutations, which would result in zero enzyme activity, are conspicuously absent; and the only nonsense mutation (G6PD Georgia; supplemental Table 2), which would do the same, has been found in a heterozygote. These data support the notion that complete loss of G6PD activity is lethal. Some variants have 2 mutations in cis: the best known is G6PD A-, in which a mutation in codon 126 (N126D, not causing G6PD deficiency), coexists with 1 of 3 other mutations (M68V, R227L, L323P). It is the combination of the 2 mutations that causes G6PD deficiency.94 Note: the term variant is currently often used to designate proteins resulting from mutations that may or may not have pathological implications. Algorithms predicting pathogenicity of variants have become popular, but, in the case of G6PD, the finding of enzyme deficiency is the best test because every enzyme-deficient variant has the potential to cause clinical manifestation.
Genotype-phenotype correlations
Given the large number of point mutations identified within the G6PD gene, we should try to understand why and how they give a particular phenotype. First, we must consider what will cause G6PD enzyme activity to be deficient. A low rate of G6PD protein synthesis is unlikely, as no regulatory mutations have been found. In principle, we might be dealing therefore with decreased stability or decreased catalytic activity (kcat). Either or both occur in different cases (see examples in Table 2), but decreased stability (that may imply impaired folding or impaired stability of dimer or tetramer) is more prominent in more cases; this is not surprising, in view of the long lifespan of red cells, whereby the physiological decrease in G6PD activity as red cells age is greatly accelerated. If instability is moderate, there will be enough G6PD for the steady-state requirements of red cells, and these will fail (ie, undergo hemolysis) only when an exogenous oxidative stress is applied. If instability is severe, G6PD activity will be insufficient to enable normal red cell survival even in the absence of oxidative stress: this will give the CNSHA (class I) phenotype.
From the evolutionary point of view, G6PD-deficient variants are enriched in amino acid residues of intermediate conservation (supplemental Figure 1 and Notaro et al24 ). We presume that mutations in more highly conserved residues are more likely to be lethal, and mutations in nonconserved residues are less likely to cause G6PD deficiency. The next question is why, once a mutation gives G6PD deficiency, the phenotype is mild (class II or III; Table 3) or severe (class I; CNSHA). The most obvious factor must be the level of residual G6PD activity: in this respect, there is a striking density of class I mutations in exon 10 (Figure 5), which comprises many of the amino acids involved in the dimer interface (failure of dimerization entails the maximum of instability); amino acid replacements near the structural NADP site (Figure 6A) or the dimer-dimer interface95 may also affect stability. Another factor is substrate affinity, because the intraerythrocytic concentrations of G6P and NADP are much below saturation96 . Therefore, a high KmG6P will be generally an adverse feature (Table 2), whereas a low KmG6P will be an advantage in the steady state (less so under oxidative stress: it might be the reason why G6PD Mediterranean, for instance, is in class II rather than in class I). The relative roles of kcat and of enzyme instability in G6PD variants have been recently explored78 and subjected to rigorous principal component analysis.81
The regulation of G6PD transcription has been investigated in some detail. The G6PD core promoter region includes 7 guanine-cytosine (GC) boxes, 2 of which, through binding of the Sp1 and AP-2 transcription factors, are essential.97 G6PD expression requires a specific chromatin conformation, as histone deacetylase inhibitors, through enhanced recruitment of Sp1 and of RNA polymerase II, enhance G6PD transcription.98
Animal models of G6PD deficiency have been reviewed elsewhere.38 Although models exist in mouse and in zebrafish, they have not yet been adopted for routine safety testing of new drugs.
Correction of G6PD deficiency
By transduction of hematopoietic stem cells with a retroviral vector harboring the human G6PD complementary DNA (cDNA), stable expression of human G6PD was obtained in primary and secondary recipient syngeneic mice.99 A similar vector was also competent for human G6PD expression in Macaque monkeys.100 In principle, severe CNSHA due to G6PD deficiency could be treated by allogeneic bone marrow transplantation or by gene therapy, but this has not yet been attempted.
In view of the fact that histone deacetylase inhibitors selectively increase the synthesis of G6PD (but not of 16 other red cell enzymes that have been tested98 ), butyrate or valproate might correct G6PD deficiency, but chronic administration would be required (or, in theory, one might contemplate using 1 of these agents to prevent or curb hemolysis under special circumstances). Very recently, high-throughput screening has been used to identify a small molecule, AG1 (2,2′-disulfanedylbis-N-(2-(1H-indol-3-yl)ethyl)ethan-1-amine), which is able to activate G6PD101 . Although the activation is less than twofold, AG1 might be a lead compound for finding more effective activators.
G6PD deficiency and malaria
A close geographic correlation between the frequency of G6PD deficiency and malaria endemicity, reminiscent of what is well known for Hb S, has been observed in very many studies (Box 3 and Figure 1); as a result, the coexistence of the 2 abnormalities is not infrequent, and it was natural to wonder whether G6PD deficiency makes sickle cell anemia worse. Overall, the effect is small.115,116 When G6PD-deficient red cells are infected by Plasmodium falciparum, they are sensed by macrophages as abnormal at an early stage; they are removed,109 which seems a highly plausible protective mechanism. The original notion that protection is a prerogative of heterozygous females,107 as in classic balanced polymorphisms, has been confirmed by several recent studies.117,118 Perhaps G6PD deficiency protects against cerebral malaria but not against malaria with severe anemia,119 although this finding has been challenged as possibly due to “collider bias.”120 It remains counterintuitive that having only a portion of G6PD-deficient red cells (as in heterozygous females) is more protective than having all G6PD-deficient red cells (as in hemizygous males). A convincing mechanistic explanation is still needed.
G6PD deficiency and malaria
1960. Based on the close geographic correlation between the frequency of G6PD deficiency and the endemicity of P falciparum malaria, Allison102 and Motulsky103 independently suggested that the latter had been a factor in Darwinian selection of the former.
1966. By micromapping the frequency of G6PD deficiency within the island of Siniscalco et al104 found a direct correlation with former malaria prevalence (by that time malaria had been eradicated in that island).
1967. By studying children with high P falciparum parasitemia in Nigeria Gilles et al105 provided evidence that G6PD deficiency may be protective against severe malaria.
1969. In patients with P falciparum malaria it is found by cytochemical analysis that parasites are present preferentially, within the same patient, in G6PD normal vs G6PD-deficient red cells.106
1972. In a study of 699 children admitted to hospital with fever it was found that the distribution of parasitemia was significantly shifted to lower levels in girls heterozygous for G6PD deficiency but not in G6PD-deficient males.107
1983-98. From in vitro cultures of P falciparum it was initially reported that parasite growth was decreased in G6PD-deficient host red cells compared with G6PD normal host red cells108 ; later it was found that under optimal conditions the growth was very similar. However, when exposed to autologous monocytes, P falciparum–infected red cells were phagocytosed at a much earlier stage of parasite development when they were G6PD-deficient than when they were G6PD normal.109
1994. Identification and cloning of the G6PD gene from P falciparum.110 This gene has sequence homology to all other known G6PD genes from other organisms,24 but it is unique by being some 300 codons longer. It was subsequently discovered that this longer gene encodes a bifunctional enzyme that has not only G6PD activity but also the metabolically closely related 6-phosphoglucono-lactonase activity.111
1995. From the combined data of a study carried out in children in Gambia and in Kenya, Ruwende et al112 reported protection against severe malaria of both G6PD-deficient males and females heterozygous for G6PD deficiency. This paper had a huge impact in this field (over 600 citations): however, years later it was invalidated by the finding that the DNA methodology used for G6PD typing must have misclassified a large proportion of the samples from the Gambia.113,114
2013-2018. Several studies have confirmed the original finding107 that protection against severe malaria (a proxy for malaria mortality) is a prerogative of heterozygotes. Considering different forms of severe malaria, protection may be greater against severe malarial anemia than against cerebral malaria.
Darwinian selection is not the only link between malaria and G6PD deficiency, as the latter was originally discovered through the study of AHA caused by exposure to primaquine (Table 1), the only drug, until recently, that effectively eliminates the dormant liver forms (hypnozoites) of Plasmodium vivax. This requires a 14 day-course of primaquine (0.75 mg/kg per day), which will regularly cause hemolysis in a G6PD-deficient person, including the majority of heterozygous females. A much lower dose (0.25 mg/kg once only) is recommended to eliminate gametocytes (after a P falciparum malaria attack has been treated with a standard course of an artemisinin combination): this is safe for G6PD-deficient persons.121 The primaquine analog tafenoquine (Table 1) also causes AHA in G6PD-deficient persons122 : we may surmise that damage to the parasite and damage to G6PD-deficient red cells go hand in hand because they are both mediated by the ability of these 4-aminoquinolines to produce ROS. Tafenoquine is attractive because, due to its longer in vivo half-life, a single dose is sufficient; however, this becomes a liability in a G6PD-deficient person, as the drug cannot be discontinued if and when signs and symptoms of AHA develop. In view of this, tafenoquine123 has been licensed with a label that positively prescribes testing for G6PD deficiency: this in turn has been a strong stimulus to the development of simple and reliable point-of-care tests for G6PD deficiency, including heterozygous females (Box 2).
Extraerythrocytic manifestations of G6PD deficiency
Because G6PD is ubiquitously expressed, one might expect manifestations of G6PD deficiency not to be limited to red cells, but rather to occur in other cells or tissues as well. Overall, these are not very prominent, mainly because, as stated herein, in most cases the molecular basis of G6PD deficiency is enzyme instability, and presumably nucleated cells can compensate for instability, if need be, through increased G6PD synthesis. The consequence of G6PD deficiency in granulocytes has been best characterized in some of the patients who have CNSHA (for example, G6PD Barcelona124 ), who suffered severe bacterial infections. The likely explanation is that granulocytes with severe G6PD deficiency are unable to produce enough NADPH to provide an effective oxidative burst (so much so that, a long time ago, G6PD deficiency had been confused with X-linked chronic granulomatous disease). A possibly related finding is the increased frequency of severe sepsis in G6PD-deficient patients after major trauma125 : interestingly, this was observed in patients with the common variant G6PD A−. There have been isolated case reports in G6PD-deficient persons of acute rhabdomyolysis with myoglobinuria and sometimes acute renal failure.126 This is a very rare occurrence when compared with AHA, with which it is usually but not always associated.127
The cells of the eye lens share with red cells loss of organelles, including the nucleus. There has been controversy as to whether G6PD deficiency favors cataract formation, but this does not seem to be the case.128 There is extensive literature on G6PD and cancer, and the results have not been uniform. A recent study has found that adenomatous polyps and colon cancer are less frequent in persons with G6PD deficiency/129 we think it is preferable to await confirmation before speculating on how G6PD deficiency might be protective. The risk of cardiovascular disease has been reported to be somewhat higher (odds ratio, 1.39; confidence interval, 1.04-1.87) in G6PD-deficient males compared with controls.130
Concluding remarks
G6PD deficiency is not a disease (except for the very small minority of patients who have CNSHA): it is a widespread genetic trait that can protect heterozygotes from dying of malaria. Yet, AHA in a G6PD-deficient child or adult is a medical emergency that, if not promptly and appropriately treated, can be fatal. On a worldwide basis, the commonest trigger of AHA is the ingestion of fava beans: favism is seen in at least 35 countries, and there are probably thousands of cases every year.32 The next common trigger, in the same and in other countries, is iatrogenic: deaths have been reported with both primaquine131 and rasburicase,132 and these deaths are preventable. Favism is preventable by population screening and health education133 ; possibly also by the introduction of fava bean cultivars with absent or very low levels of vicine and convicine.134
As hematologists, we have learnt much from G6PD-deficient patients. At the same time, the biology of G6PD is eminently interdisciplinary, having been a model system in biochemical genetics and in understanding how the red cell responds to oxidative attack; a tool for studying X-chromosome inactivation (the most spectacular epigenetic event in human development); a tool, for years, for studying clonal populations; a pioneer in the molecular genetics of enzymopathies; and the best characterized example in humans of an X-linked genetic polymorphism balanced by Darwinian selection exerted by malaria.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank all of their collaborators in Ibadan, Naples, London, New York, Genova, Firenze, Bangkok, Ouagadougou, Dar es Salaam, and elsewhere, and particularly José Bautista for careful review of the manuscript.
This paper is dedicated to the memory of Olaniyi Babalola and Giorgio Battistuzzi, who did early studies on the biochemistry and genetics of G6PD variants, and of Graziella Persico, who cloned the human G6PD gene.
Authorship
Contribution: All authors contributed to writing the paper and to assembling the large amount of supplementary materials.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Lucio Luzzatto, Hematology, Muhimbili University of Health and Allied Sciences, PO Box 65001, Dar es Salaam, United Republic of Tanzania; e-mail: lluzzatto@blood.ac.tz.
