


Abstract
Congenital dyserythropoietic anemias (CDAs) are a heterogeneous group of inherited anemias that affect the normal differentiation–proliferation pathways of the erythroid lineage. They belong to the wide group of ineffective erythropoiesis conditions that mainly result in monolinear cytopenia. CDAs are classified into the 3 major types (I, II, III), plus the transcription factor-related CDAs, and the CDA variants, on the basis of the distinctive morphological, clinical, and genetic features. Next-generation sequencing has revolutionized the field of diagnosis of and research into CDAs, with reduced time to diagnosis, and ameliorated differential diagnosis in terms of identification of new causative/modifier genes and polygenic conditions. The main improvements regarding CDAs have been in the study of iron metabolism in CDAII. The erythroblast-derived hormone erythroferrone specifically inhibits hepcidin production, and its role in the mediation of hepatic iron overload has been dissected out. We discuss here the most recent advances in this field regarding the molecular genetics and pathogenic mechanisms of CDAs, through an analysis of the clinical and molecular classifications, and the complications and clinical management of patients. We summarize also the main cellular and animal models developed to date and the possible future therapies.
Introduction
Congenital dyserythropoietic anemias (CDAs) belong to a wide group of conditions that are characterized by inefficient erythropoiesis that mainly result in monolinear cytopenia. For a long time, bone marrow morphological abnormalities were the key diagnostic features of CDAs, such as erythroid hyperplasia with binuclearity, or multinuclearity of late erythroblasts. Unfortunately, these features are not specific to CDAs because they can be present in other acquired conditions that involve erythropoietic stress, such as iron deficiency and preterm birth. This aspect might account for the difficulties in the diagnosis of CDAs. Nevertheless, the working classification of the 3 major types of CDAs (I, II, III) remains based on the morphological features of erythroblasts in the bone marrow. As this classification has been extensively examined previously,1-3 it will not be discussed in detail here. We focus instead on the most recent advances in the genetics and pathogenic mechanisms of CDAs.
Clinical and molecular classification of CDAs
CDAs encompass a vast group of hypoproliferative anemias, which can be classified into 5 types: CDA types I, II, and III, transcription-factor-related CDAs, and the CDA variants.
CDA type I
CDA type I (CDAI) is characterized by severe or moderate anemia, which is generally macrocytic, and relative reticulocytopenia and congenital anomalies, such as skeletal abnormalities, chest deformity, and short stature.4 Distal limb anomalies are well documented for ∼10% of these patients, although they have been described also in other CDA subtypes.5 In the bone marrow, 2.4% to 10% of late erythroblasts are binucleate, and most of these have nuclei at different stages of erythroid differentiation.3,6 However, the typical morphological feature of CDAI is the presence of thin internuclear chromatin bridges between the nuclei pairs of intermediate erythroblasts (1% to 8% of cells examined) (Figure 1A).6 Indeed, internuclear chromatin and cytoplasmic bridges have been observed in 79% of patients with CDAI.7 Under electron microscopy, their heterochromatin is denser than normal and forms demarcated clumps with small translucent vacuoles, which gives rise to the metaphor of the classical “spongy” or “Swiss cheese” appearance of the nucleus.6,7
![Morphological and molecular features of patients with CDA. (A) Light microscopy analysis of the bone marrow from patients with different CDA subtypes. CDA patients generally show erythroid hyperplasia. Red arrows indicate typical findings for each CDA subtype: CDAI, internuclear chromatin bridging; CDAII, binucleate erythroid precursors; CDAIII, giant multinucleated erythroblasts; CDAIV, multinucleate erythroblasts; and CAD deficiency, binucleate CDAII-like precursors. (B) Pie chart showing the frequencies of the different CDA subtypes diagnosed after genetic testing in patients clinically suspected of having CDA. The frequency of each condition was calculated as the ratio between the number of patients in each CDA subtype and the overall count of patients tested (n = 218 patients [those included in our international registry of CDAs from 1995 to 2019]). Six patients originally suspected of CDA showed conclusive diagnosis of acquired dyserythropoiesis: 2 patients with liver failure, 2 with iron-deficiency anemia, 1 with erythrophagocytosis, and 1 with transient erythroblastopenia. Syndromic CDA refers to 1 patient with a mutation in the CAD gene. GATA1-related cytopenias include: X-linked thrombocytopenia with or without dyserythropoietic anemia; congenital erythropoietic porphyria; and idiopathic cytopenias of undetermined significance. Other hereditary anemias (HA) include: hereditary spherocytosis; hereditary dehydrated stomatocytosis; red cell enzymatic defects; and sideroblastic anemia. The undiagnosed cases were evaluated by analysis of the CDA gene panel, by extended targeted next-generation sequencing for hereditary anemias, or by whole-exome sequencing. (C) Bubble chart defining the lengths of the coding sequences of each CDA-causative gene and their relative pathogenicity scores. These scores were calculated by combining the constraint metrics of each gene available at the ExAC database (http://exac.broadinstitute.org/). High pathogenicity scores identify increased constraints (intolerance to variation). The more intolerant to variation a gene is, the less likely it is to be mutated. The size of each bubble represents the frequency of the mutations in each gene, as calculated by the ratio of the number of mutated alleles for each gene and the overall count of disease alleles (n = 149, from 78 patients included in our international registry of CDAs from 2008 to 2019).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/11/10.1182_blood.2019000948/1/m_bloodbld2019000948cf1.png?Expires=1769081171&Signature=41PKI9NgfLrNwYbR9fnFsWjZXqjuNKtghm1zcpT2Q~GEgsfUd7vd-bFzai2flpfwow6fcawCR7sxgllfb60izF264JsRwyslHCs~FdN06jYC8MAOX0IxUHxcVHy1d2GkT5hDfc8N~EtnvSi0TI-rehBaUGqMu2ymkWLrz~U3ilmODPHoJq1TtA4s4R5VlPbfeIJBuPlTgkLN8XSiXxuX-OQCYmphEma7lZa70nknwUHGvD7ahActPa4MWsVLyg2sw~hSjrwHVul0sAvPuhSOHdI30emJc5zhQYqjMfLNs3wbVH7fKQn4haDp7gC~EEdjO6dEgg75fHGGBMWso84waw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Morphological and molecular features of patients with CDA. (A) Light microscopy analysis of the bone marrow from patients with different CDA subtypes. CDA patients generally show erythroid hyperplasia. Red arrows indicate typical findings for each CDA subtype: CDAI, internuclear chromatin bridging; CDAII, binucleate erythroid precursors; CDAIII, giant multinucleated erythroblasts; CDAIV, multinucleate erythroblasts; and CAD deficiency, binucleate CDAII-like precursors. (B) Pie chart showing the frequencies of the different CDA subtypes diagnosed after genetic testing in patients clinically suspected of having CDA. The frequency of each condition was calculated as the ratio between the number of patients in each CDA subtype and the overall count of patients tested (n = 218 patients [those included in our international registry of CDAs from 1995 to 2019]). Six patients originally suspected of CDA showed conclusive diagnosis of acquired dyserythropoiesis: 2 patients with liver failure, 2 with iron-deficiency anemia, 1 with erythrophagocytosis, and 1 with transient erythroblastopenia. Syndromic CDA refers to 1 patient with a mutation in the CAD gene. GATA1-related cytopenias include: X-linked thrombocytopenia with or without dyserythropoietic anemia; congenital erythropoietic porphyria; and idiopathic cytopenias of undetermined significance. Other hereditary anemias (HA) include: hereditary spherocytosis; hereditary dehydrated stomatocytosis; red cell enzymatic defects; and sideroblastic anemia. The undiagnosed cases were evaluated by analysis of the CDA gene panel, by extended targeted next-generation sequencing for hereditary anemias, or by whole-exome sequencing. (C) Bubble chart defining the lengths of the coding sequences of each CDA-causative gene and their relative pathogenicity scores. These scores were calculated by combining the constraint metrics of each gene available at the ExAC database (http://exac.broadinstitute.org/). High pathogenicity scores identify increased constraints (intolerance to variation). The more intolerant to variation a gene is, the less likely it is to be mutated. The size of each bubble represents the frequency of the mutations in each gene, as calculated by the ratio of the number of mutated alleles for each gene and the overall count of disease alleles (n = 149, from 78 patients included in our international registry of CDAs from 2008 to 2019).
Morphological and molecular features of patients with CDA. (A) Light microscopy analysis of the bone marrow from patients with different CDA subtypes. CDA patients generally show erythroid hyperplasia. Red arrows indicate typical findings for each CDA subtype: CDAI, internuclear chromatin bridging; CDAII, binucleate erythroid precursors; CDAIII, giant multinucleated erythroblasts; CDAIV, multinucleate erythroblasts; and CAD deficiency, binucleate CDAII-like precursors. (B) Pie chart showing the frequencies of the different CDA subtypes diagnosed after genetic testing in patients clinically suspected of having CDA. The frequency of each condition was calculated as the ratio between the number of patients in each CDA subtype and the overall count of patients tested (n = 218 patients [those included in our international registry of CDAs from 1995 to 2019]). Six patients originally suspected of CDA showed conclusive diagnosis of acquired dyserythropoiesis: 2 patients with liver failure, 2 with iron-deficiency anemia, 1 with erythrophagocytosis, and 1 with transient erythroblastopenia. Syndromic CDA refers to 1 patient with a mutation in the CAD gene. GATA1-related cytopenias include: X-linked thrombocytopenia with or without dyserythropoietic anemia; congenital erythropoietic porphyria; and idiopathic cytopenias of undetermined significance. Other hereditary anemias (HA) include: hereditary spherocytosis; hereditary dehydrated stomatocytosis; red cell enzymatic defects; and sideroblastic anemia. The undiagnosed cases were evaluated by analysis of the CDA gene panel, by extended targeted next-generation sequencing for hereditary anemias, or by whole-exome sequencing. (C) Bubble chart defining the lengths of the coding sequences of each CDA-causative gene and their relative pathogenicity scores. These scores were calculated by combining the constraint metrics of each gene available at the ExAC database (http://exac.broadinstitute.org/). High pathogenicity scores identify increased constraints (intolerance to variation). The more intolerant to variation a gene is, the less likely it is to be mutated. The size of each bubble represents the frequency of the mutations in each gene, as calculated by the ratio of the number of mutated alleles for each gene and the overall count of disease alleles (n = 149, from 78 patients included in our international registry of CDAs from 2008 to 2019).
CDAI is inherited as an autosomal recessive disorder that is caused by biallelic mutations in 2 different loci that account for 90% of CDAI cases: CDAN1 and C15orf41 (Figure 1B-C).4 To date, 51 causative mutations in CDAN1 and 6 in C15orf41 have been documented.4,8-10 Both of the proteins encoded by these 2 genes are thus likely to have critical roles in DNA repair and/or chromatin reassembly following DNA replication (Figure 1).3,4 CDAN1 (15q15.2) was the first causative gene to be identified (OMIM #224120),11 and it encodes a cell cycle-regulated protein, Codanin-1,12 which acts in nucleosome assembly and disassembly through formation of the cytosolic Asf1–H3–H4–importin-4 complex.13 The second locus associated with CDAI, C15orf41 (15q14) (OMIM #615631), is an uncharacterized gene that is predicted to encode a divalent metal-ion-dependent restriction endonuclease with homology to the Holliday junction resolvases.8 Like Codanin-1, C15orf41 has also been suggested to interact with Asf1b.14 Accordingly, the C15orf41 protein localizes either to the cytosol or (mainly) to the nucleus, which suggests a dual function of this protein within these 2 subcellular compartments (Figure 2).10 Of note, Codanin‐1 and C15orf41 are widely expressed, and loss of either of these 2 proteins is incompatible with life, although their alterations mainly affect the erythroid lineage. A possible explanation is that erythroid progenitors have a uniquely fast cell cycle, whereas another hypothesis includes nuclear extrusion in erythroblasts, which requires the eviction of histones, such as H3 and H4; both of these proteins might have a role in this process.4
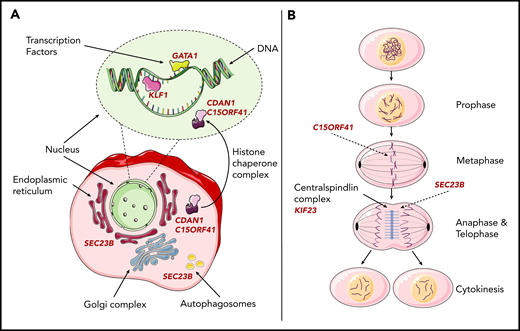
Schematic representation of the pathogenic mechanisms of CDAs at the subcellular level. (A) Pathogenic mechanisms of CDAs I and II, and of the transcription-factor-related CDAs at the erythroblast level, with schematic representation of erythroblasts and the roles of the causative proteins in the different subcellular compartments. SEC23B is a COPII component that is involved in anterograde transport from the endoplasmic reticulum toward the Golgi compartment. Moreover, SEC23B is involved in autophagy. The pathogenic mechanisms of transcription factor-related CDAs and CDAIa and Ib arise from impairment of mechanisms involved in DNA synthesis and chromatin assembly. During cell division, GATA1 and KLF1 might be retained focally within mitotic chromatin to facilitate the rapid reactivation of the transcription of tissue-specific genes upon entry into G1; codanin-1 and C15orf41 interact with the cytosolic Asf1–H3–H4–importin-4 complex that is involved in nucleosome assembly and disassembly. (B) The pathogenic mechanisms of CDAs I, II, and III at the erythroblast level, with schematic representation of cell division and the roles of the causative proteins in the different mitotic stages. C15orf41 encodes a protein with homology to the Holliday junction resolvases, which are involved in chromosome segregation. The KIF23 mutant results in furrow regression, and thus inhibits cytokinesis. SEC23B has a crucial role in the assembly or deconstruction of the midbody during cytokinesis.
Schematic representation of the pathogenic mechanisms of CDAs at the subcellular level. (A) Pathogenic mechanisms of CDAs I and II, and of the transcription-factor-related CDAs at the erythroblast level, with schematic representation of erythroblasts and the roles of the causative proteins in the different subcellular compartments. SEC23B is a COPII component that is involved in anterograde transport from the endoplasmic reticulum toward the Golgi compartment. Moreover, SEC23B is involved in autophagy. The pathogenic mechanisms of transcription factor-related CDAs and CDAIa and Ib arise from impairment of mechanisms involved in DNA synthesis and chromatin assembly. During cell division, GATA1 and KLF1 might be retained focally within mitotic chromatin to facilitate the rapid reactivation of the transcription of tissue-specific genes upon entry into G1; codanin-1 and C15orf41 interact with the cytosolic Asf1–H3–H4–importin-4 complex that is involved in nucleosome assembly and disassembly. (B) The pathogenic mechanisms of CDAs I, II, and III at the erythroblast level, with schematic representation of cell division and the roles of the causative proteins in the different mitotic stages. C15orf41 encodes a protein with homology to the Holliday junction resolvases, which are involved in chromosome segregation. The KIF23 mutant results in furrow regression, and thus inhibits cytokinesis. SEC23B has a crucial role in the assembly or deconstruction of the midbody during cytokinesis.
CDA type II
CDA type II (CDAII) (OMIM #224100) is the most common form among the CDAs (Figure 2).15 Clinically, this condition is characterized by normocytic anemia of variable degrees, with normal or slightly increased reticulocyte counts. CDAII is often accompanied by jaundice and splenomegaly because of the hemolytic component. Generally, these patients are mildly affected, although a wide spectrum of clinical presentations is known.16 Indeed, ∼10% of cases are symptomless, whereas 20% of patients are transfusion dependent.17,18 Like CDAI, the bone marrow in CDAII is hypercellular, with distinct erythroid hyperplasia. In CDAII, the most specific finding is the presence of binucleate cells with 2 nuclei at the same erythroid maturation stage (Figure 1A). If >10% binucleate erythroblasts are seen together with >2% of the cells with karyorrhexis (ie, a fragmentation of the nucleus with breakup of the chromatin into unstructured granules), the diagnosis of CDAII is almost certain.6 Under electron microscopy, the mature erythroblasts show a discontinuous double membrane, which is due to the presence of vesicles loaded with proteins of the endoplasmic reticulum that appear to be positioned beneath the plasma membrane.3 A pathognomonic biochemical feature of the membrane proteins isolated from CDAII erythrocytes is the presence of hypoglycosylated band 3, which is observed in 95% of patients with CDAII.17
CDAII is inherited as an autosomal recessive disorder that is due to biallelic mutations in the SEC23B gene (20p11.23).19,20 This is the most mutated locus among the CDA causative genes (Figure 1B-C). Indeed, ∼100 pathogenic variants have been identified so far, most of which are inherited as private mutations, even if recurrent variants have been described in different geographic regions.17,21-24 SEC23B encodes the homonymous member of the cytoplasmic coat protein II (COPII) complex, which is involved in intracellular vesicle trafficking in eukaryotic cells. In particular, it is a GTPase-activating protein important for regulation of the coating process.25 Despite the ubiquitous expression of SEC23B, the effects of mutations in this gene are confined to the erythroid lineage, and the basis of this erythroid specificity has been essentially defined.19,26 As detailed later, it is difficult to establish the pathophysiology of CDAII, mainly because of the absence of a reliable animal model. However, some hypotheses have been addressed to date (Figure 2). First, the erythroid specificity of the phenotype might be related to either the tissue-specific expression of SEC23B during erythroid differentiation in human19,26 or the need for erythroid-specific cargoes (such as band 3), which might require high levels and full function of a specific COPII component to be correctly transported.25 Second, SEC23B might have an active role in the assembly or deconstruction of the midbody, and its loss of function might account for the impaired cytokinesis observed in CDAII erythroblasts.3 Finally, a recent study described the involvement of SEC23B in autophagy. In particular, it was shown that the protein FBXW5 targets SEC23B for proteasomal degradation, and that this event limits the autophagic flux in the presence of nutrients. Conversely, in response to starvation, ULK1 phosphorylates SEC23B, which prevents its interaction with FBXW5, and therefore inhibits its degradation. Phosphorylated and stabilized SEC23B associates with 2 other COPII components, SEC24A and SEC24B, to promote autophagic flux.27 This interesting study by Jeong et al27 opened the way to dissect the role of SEC23B in the pathogenic mechanism of CDAII through interference with the autophagic process to discover new therapeutic targets. Moreover, this is in line with previous studies that have demonstrated the requirement for COPII proteins in autophagy, as well as the importance of autophagy in the final stage of reticulocyte maturation.28,29
CDA type III
CDA type III (CDAIII) (OMIM #105600) is the rarest of these 3 classical CDA forms.2 Most of these patients described to date belong to 2 unrelated families, 1 American and the other Swedish, who show an autosomal dominant inheritance pattern.28 The patients show absent or moderate anemia, with normal or slightly increased mean corpuscular volume, slight relative reticulocytopenia, jaundice, and signs of hemolysis. Splenomegaly is usually absent. Light microscopy of the bone marrow highlights erythroid hyperplasia, with characteristic giant multinucleate erythroblasts (Figure 1A). Under electron microscopy, clefts within heterochromatin, autophagic vacuoles, iron-laden mitochondria, and myelin figures in the cytoplasm of the erythroblasts have been described. A large proportion of patients in the Swedish family also show visual disturbances, with macular degeneration, angioid streaks, and monoclonal gammopathy; some have developed multiple myeloma.30
CDAIII was the first CDA to be described (in 1951), although its genetic etiology was only assessed more than 60 years later.30,31 A unique causative variant c.2747C>G (p.P916R) in the KIF23 gene (15q21) has been identified to date in both the American and Swedish pedigrees. This gene encodes mitotic kinesin-like protein 1, which is a key component of the central mitotic spindle, a subcellular structure that ensures faithful separation of the cells during late mitosis (Figure 2).30 The mitotic kinesin-like protein 1 mutation affects the function of this protein during cytokinesis, which leads to the formation of large multinucleate erythroblasts in the bone marrow of these patients.30
Transcription factor-related CDA
This subgroup includes CDA type IV (CDAIV) (OMIM #613673) and X-linked thrombocytopenia with or without dyserythropoietic anemia (XLTDA) (OMIM #300367). To date, 8 patients with CDAIV have been reported.32-38 They have hemolytic anemia, which is generally severe, with normal or slightly increased reticulocyte count, and markedly elevated fetal hemoglobin levels. Hypercellular bone marrow and binucleate or multinucleate erythroblasts are observed. Electron microscopy shows immature erythroid progenitors with atypical cytoplasmic inclusions, invagination of the nuclear membrane, and marked heterochromatin.32,33
CDAIV is an autosomal dominant condition caused by a unique heterozygous variant c.973G>A (p.E325K) in the KLF1 gene (19p13.2) (Figure 2). This gene encodes the homonymous erythroid-specific transcription factor KLF1, which has a critical role in the regulation of the switch between fetal and adult hemoglobin expression, and is required in terminal erythroid differentiation.39 Recently, the molecular mechanism of the KLF1-E325K mutation has been investigated.38,40 These studies have demonstrated that the erythroid cells of these patients show dysregulation of global expression of erythroid, cell-surface, membrane-transport, cytokinesis, iron-utilization, and cell-cycle regulator genes. The KLF1-E325K mutation results in multifactorial interactions with target DNA sequences (ie, dominant-negative effects, dominant-positive effects, or haplo-insufficiency).38 The main effect of the KLF1-E325K mutation is either impaired recognition by this mutated KLF1 of its normal cognate site, or incorrect protein complex formation that interferes with the wild-type activity at target sites.40
XLTDA belongs to a group of clinically heterogeneous conditions characterized by macrothrombocytopenia with hypogranulated platelets, bleeding tendency, and mild to severe anemia. At the extreme end of the clinical spectrum, severe hemorrhage and/or erythrocyte transfusion dependence are lifelong; at the milder end, anemia and the risk of bleeding can decrease spontaneously with age.41 Bone marrow features are dyserythropoiesis and impaired megakaryopoiesis (ie, megakaryocytes are reduced in number and are aberrant, with cytoplasmic vacuoles and an absence of platelet membrane demarcation).
XLTDA is caused by mutations in GATA1 (Xp11.23) (Figure 2), an X-linked gene that encodes a DNA-binding protein with 2 zinc fingers and a transactivation domain. GATA1 has an essential role in development and maintenance of both erythroid and megakaryocytic lineages.42 GATA1 mutations in benign hematological disorders often affect the function of the N-terminal zinc finger, which mediates interactions with the cofactor “friend of GATA1.”42 Because of the X-linked inheritance, males are predominantly affected, and the severity and specificity of the phenotype depends on the imbalance in GATA1 function, which is determined by the mutation type and its expression level.41 However, female mutation carriers can show borderline hemoglobin levels and platelet counts that range from normal to severely reduced, which reflects the skewed X chromosome inactivation. Of note, increased bleeding with increased age has been reported in elderly females, which is probably related to age-dependent skewing of the X chromosome inactivation and to oligoclonal hematopoiesis.43
Because of the roles of transcriptional regulators of different genes and pathways, several mutations identified in KLF1 or GATA1 result in heterogeneous phenotypes. Indeed, haploinsufficient mutations in KLF1 account for the in(Lu) blood type, hereditary persistence of fetal hemoglobin, CDAIV, and mild thalassemia. Similarly, GATA1-related cytopenias are hallmarked by thrombocytopenia that can be associated to impaired erythropoiesis, dyserythropoietic anemia, congenital erythropoietic porphyria, thalassemia, or Diamond Blackfan anemia-like disease.3 Moreover, genetic variants in KLF1 and GATA1 might also have roles as modifiers of the phenotype. Indeed, coinheritance of pathogenic variants in KLF1/GATA1 and in other relevant erythrocyte genes has been reported in some cases that show a severe clinical course.36,44,45 Additionally, hypomorphic polymorphism of GATA1 has been described recently, which exacerbates the phenotype induced by mutations in GATA1-dependent genes.46
CDA variants
CDA variants show either isolated or syndromic CDA-like conditions. For instance, an X-linked dominant macrocytic dyserythropoietic anemia with iron overload has been described in multiple female individuals.37,47,48 This condition is related to heterozygous mutations in the ALAS2 gene (Xp11.21), which is already known to be the causative gene of X-linked sideroblastic anemia in males.
Among the syndromic conditions, Majeed syndrome (OMIM #609628) is a rare autosomal recessive disorder that is hallmarked by chronic recurrent multifocal osteomyelitis, inflammatory dermatosis, and hypochromic microcytic anemia with dyserythropoiesis.49 The causative gene is LPIN2 (18p11.31), which encodes a phosphatidate phosphatase important in lipid metabolism.50,51
More recently, a syndromic condition has been described that comprises severe neurodegenerative disease, which is associated with mild CDA-like anemia with marked anisopoikilocytosis and abnormal glycosylation of the erythrocyte proteins band-3 and RhAG.37,52,53 This disorder, known as early infantile epileptic encephalopathy-50 (OMIM #616457), is caused by biallelic mutations in the CAD gene (2p23.3), which encodes a trifunctional enzyme that catalyzes the first steps of de novo pyrimidine biosynthesis. Interestingly, as pyrimidines can be recycled from uridine, the administration of uridine has been shown to ameliorate the phenotype of these CAD-deficient patients.52,53
A unique study described 2 consanguineous families with dyserythropoiesis associated with exocrine pancreatic insufficiency and calvarial hyperostosis resulting from a homozygous mutation in the COX4I2 gene (20q11.21), which encodes a subunit of cytochrome c oxidase, the terminal enzyme in the respiratory chain.54 The COX4I2 gene localizes in close proximity to the original linkage region of the CDAII causative gene that subsequently changes.55 Thus, cosegregation of 2 different conditions in these patients cannot be excluded because SEC23B mutations have not been investigated.
Finally, a case of mevalonate kinase deficiency associated to morphological erythroblast abnormalities like CDAII has also been reported, which is caused by compound heterozygosity for 2 missense mutations in the MVK gene (12q24.11).56
Recently, through the widespread use of new technologies for DNA sequencing, some new genes have been suggested as causative of unspecific forms of CDAs. For example, biallelic mutations in the PARP4 gene that encodes a poly-ADP ribose polymerase have been identified in 2 siblings with a CDAII-like anemia.57 A de novo missense variant in the VPS4A gene that encodes an ATPase that appears to have a critical role in cytokinesis has been reported for a patient with CDAI-like anemia.58 Similarly, a heterozygous missense variant in the PRDX2 gene that encodes the antioxidant enzyme peroxiredoxin II has been described in a family with an atypical, dominantly inherited CDA.59 However, no additional cases with PARP4, VPS4A, or PRDX2 mutations have been reported to date, and a conclusive functional validation of the pathogenicity of these identified variants is still lacking.
Cellular and animal models of CDAs
The main hurdle to advancing our understanding of the pathogenic mechanisms that underlie CDAs is the lack of appropriate models for their study. Interference with SEC23B expression in the K562 human erythroleukemic cell line by either plasmid transfection or stable lentivirus infection results in the cytokinesis defects observed in patients with CDAII. This includes the presence of binucleate cells and an increase in the proportion of cells in the G2+M phase of the cell cycle.19,60 Knock-out mice for the Sec23b gene die perinatally instead, with huge pancreatic degeneration61-63 and no features of CDAII or other signs of anemia.61,64 Murine and human tissues show different Sec23a/Sec23b expression ratio. Indeed, the expression of Sec23a, but not Sec23b, is maintained during murine terminal erythroid differentiation.26 Moreover, as observed in some CDAII patients,65 knock-down of Sec23b in mice results in a compensatory increase in the paralog gene, Sec23a.66 The knock-down of zebrafish sec23b by morpholino leads to aberrant erythrocyte development.19 On the contrary, the knock-out zebrafish model (sec23b−/−) did not survive beyond 3 weeks of age.66 This lethality that results from sec23b disruption in zebrafish can be rescued by a sec23a-expressing transgene. Accordingly, the Sec23a coding sequence inserted into the endogenous murine Sec23b locus fully rescues the mortality and severe pancreatic phenotype of Sec23b deficiency in the mouse.66
For CDAI, the only Cdan1 transgenic mouse model created to date has shown embryonic lethality.67 For the C15ORF41 gene, 2 cellular models have been created: 1 in human umbilical-derived erythroid precursor (HUDEP)-2 cells using CRISPR/CAS9 technology,68 and the other in K562 cells through overexpression of the H230P and Y94S mutations.10 The model in the HUDEP-2 cells remains to be investigated, whereas the K562 models showed impaired erythroid differentiation.10
Diagnostic workflow of CDAs
As for other hereditary anemias, the classical diagnostic workflow for CDA includes different lines of investigation, which start from the analysis of family and personal history, move on to biochemical and morphological evaluation, and end with genetic testing.1 However, the high clinical and genetic heterogeneity of these conditions often hampers correct clinical diagnosis. For instance, frequent misdiagnosis between CDAI and hereditary stomatocytosis is well known,69 as well as between CDAII and hereditary spherocytosis,3 although some screening markers have been proposed.17,70 Recently, ektacytometry has been used to differentiate between CDAII and hereditary spherocytosis, although some of the values between these conditions show overlap.71 Differential diagnosis based on a label-free optical marker has also been reported.72
Currently, the framework of diagnosis has changed (Figure 3). Genetic testing has become the frontline system for differential diagnosis of patients affected by CDAs. Molecular diagnosis is essential when the clinical diagnosis is not certain (eg, in patients who require frequent transfusions) (Figure 3). Moreover, genetic testing can be performed using small amounts of peripheral blood early in the diagnostic process, which allows correct patient management to be instituted and reduces the time between clinical suspicion and diagnosis.4,37 However, biochemical and morphological evaluations are still used when variants of uncertain significance are identified to better define their pathogenicity (Figure 3).
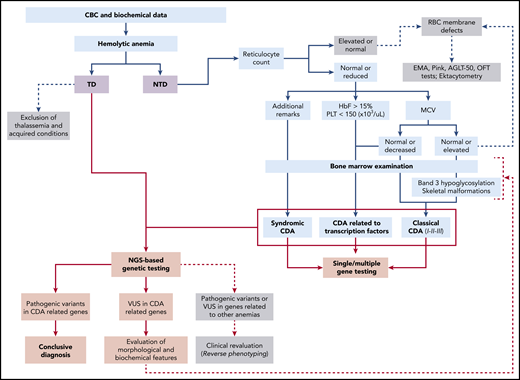
Diagnostic workflow of CDAs. Flow diagram for the differential diagnosis of CDAs and their related hereditary anemias. This illustrates the main differences between patients with transfusion-dependent (TD) anemia compared with patients with nontransfusion-dependent (NTD) anemia. Light blue shading indicates the main steps to reach diagnosis of CDAs by complete blood count (CBC) analysis and specific tests, such as examination of bone marrow, analysis of band 3 hypoglycosylation by sodium dodecyl sulfate polyacrylamide gel electrophoresis (for CDAII), examination of skeletal malformations (for CDAI), assessment of additional clinical features for syndromic conditions, and evaluation of fetal hemoglobin levels for transcription factor-related CDAs. Light red shading indicates the main steps to reach molecular diagnosis of these conditions. HbF, fetal hemoglobin; MCV, mean corpuscular volume; PLT, platelet count; Ret count, reticulocyte count; RBC, red blood cell; t-NGS, targeted next-generation sequencing; VUS, variant of uncertain significance; WES, whole-exome sequencing; XLTDA, X-linked thrombocytopenia with or without dyserythropoietic anemia.
Diagnostic workflow of CDAs. Flow diagram for the differential diagnosis of CDAs and their related hereditary anemias. This illustrates the main differences between patients with transfusion-dependent (TD) anemia compared with patients with nontransfusion-dependent (NTD) anemia. Light blue shading indicates the main steps to reach diagnosis of CDAs by complete blood count (CBC) analysis and specific tests, such as examination of bone marrow, analysis of band 3 hypoglycosylation by sodium dodecyl sulfate polyacrylamide gel electrophoresis (for CDAII), examination of skeletal malformations (for CDAI), assessment of additional clinical features for syndromic conditions, and evaluation of fetal hemoglobin levels for transcription factor-related CDAs. Light red shading indicates the main steps to reach molecular diagnosis of these conditions. HbF, fetal hemoglobin; MCV, mean corpuscular volume; PLT, platelet count; Ret count, reticulocyte count; RBC, red blood cell; t-NGS, targeted next-generation sequencing; VUS, variant of uncertain significance; WES, whole-exome sequencing; XLTDA, X-linked thrombocytopenia with or without dyserythropoietic anemia.
Single gene testing can be still suggested for patients with complete phenotyping (at the clinical, biochemical, and morphological levels; Figure 3). However, current approaches to genetic analysis here include next-generation sequencing, using custom targeted panels and whole-exome sequencing.37,73-75 The reported target panels for hereditary anemias are composed of variable numbers of genes (eg, 50-200), and they provide diagnostic yields from 38% to 65%, depending on the number and types of genes included and the depth of the phenotypic assessment undertaken.4,37,73-75 Of note, this diagnostic approach leads to modification of the original clinical diagnosis in 10% to 40% of the patients investigated (Figure 2).37,73 A recent study of a case series showed that among the patients originally classified with CDAs, 18% had a conclusive diagnosis of dehydrated hereditary stomatocytosis, whereas 45% had a final diagnosis of chronic anemia from enzymatic defects.37 In particular, most of these patients showed mutations in PKLR, the causative gene of pyruvate kinase deficiency.37,73
Complications
The main complications of CDAs are associated with chronic hemolytic anemia: iron overload, hydrops fetalis, aplastic crisis, hyperbilirubinemia, gallstones, and splenomegaly. Iron overload is the most frequent complication of CDAs. It is mainly the result of ineffective erythropoiesis, and it is also linked to both the transfusion regimen and the hemolytic component.3 Among CDAII patients, ∼30% of those who are not transfusion dependent show increased ferritinemia (ferritin >300 ng/mL), whereas 17% of them show marked hemosiderosis (ferritin >600 ng/mL).3,17 CDA patients with severe iron overload also show liver damage (ie, cirrhosis), heart failure, diabetes mellitus, and hypergonadotropic hypogonadism (with related subfertility and osteopenia). Moreover, the severe phenotypes can be related to coinheritance of modifier mutations, such as polymorphic variants in the HFE gene, which causes hemochromatosis type 2.76
In recent years, and mainly for CDAII, the mechanism of hepatic iron overload has been dissected out. Reduced expression of the hepatic hormone hepcidin was previously shown, and some erythroid regulators have been proposed as pathological suppressors of hepcidin expression, such as growth differentiation factor 15 (GDF15),77-79 although GDF15 alone does not appear to be necessary for physiological hepcidin suppression.80 The erythroblast-derived hormone erythroferrone (ERFE),81 which specifically inhibits hepcidin production, has been studied in CDAII patients who showed increased levels of ERFE compared with healthy controls (Figure 4).60 Of note, patients with high levels of ERFE also had reduced hemoglobin levels and increased erythropoietin levels compared with patients with lower levels of ERFE. This, in turn, leads to reduced hepcidin and hepcidin-to-ferritin ratio, which results in augmented iron delivery to the erythron.60 Recently, a recurrent low-frequency variant in the ERFE gene was identified (ie, p.A260S) in 12.5% of CDAII patients with severe phenotypes, using an 81-gene targeted sequencing panel for modifier genes.82 The ERFE-A260S variant results in increased levels of ERFE, with subsequent marked impairment of iron regulation pathways at the hepatic level. Functional characterization of ERFE-A260S in the Huh7 hepatic cell system demonstrated its modifier role in iron overload through impairment of the BMP/SMAD pathway, and the consequent reduction in hepcidin expression. This finding confirms the role of ERFE in the development of hepatic iron overload in CDAII, even if other actors might be involved in this scenario to explain the impairment of iron metabolism in these patients.60,82
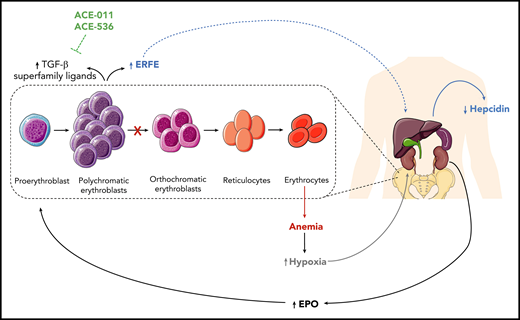
Schematic representation of the pathogenic mechanisms of CDAII at the systemic level. The pathogenic mechanism of CDAII at a systemic level, highlighting the role of erythroferrone (ERFE) in the interplay between the bone marrow and hepatic compartments. At the bone marrow level, block of erythroid maturation results in accumulation of erythroblasts that secrete ERFE. The increased levels of ERFE are responsible for the suppression of Hamp expression, which codes for hepcidin, and this can result in liver iron overload. EPO, erythropoietin; TGF-β, transforming growth factor-β.
Schematic representation of the pathogenic mechanisms of CDAII at the systemic level. The pathogenic mechanism of CDAII at a systemic level, highlighting the role of erythroferrone (ERFE) in the interplay between the bone marrow and hepatic compartments. At the bone marrow level, block of erythroid maturation results in accumulation of erythroblasts that secrete ERFE. The increased levels of ERFE are responsible for the suppression of Hamp expression, which codes for hepcidin, and this can result in liver iron overload. EPO, erythropoietin; TGF-β, transforming growth factor-β.
Clinical management of CDA patients and future therapies
The standard clinical management of CDA patients is evaluation of the complete blood count and the iron balance parameters, with monitoring every 6 months. The standard treatment of cases with severe anemia (hemoglobin, <7 g/dL) is transfusion. In cases of suspected CDA in utero, it is important to search for hydrops using prenatal ultrasound scanning, and in cases of fetal anemia, in utero transfusions are needed. Hematopoietic stem cell transplantation (HSCT) is another therapeutic option in severe cases. HSCT has been successfully performed in 2 CDAI83,84 and 7 CDAII17,85-87 patients. Among these, 2 matched unrelated donor HSCTs have been reported: both are alive, with partially resolved or no serious complications.87 Moreover, a recent study described the outcome of 39 CDA patients who underwent bone marrow transplantation.88 The outcome appears to be improved for patients transplanted from matched sibling donors and without iron overload. Of note, iron overload is an important factor that should be treated before transplantation.88
One specific treatment that is available for CDAI patients is interferon-α. Although the number of treated subjects is limited, patients with CDAN1 mutations, but not those with the C15orf41 variants, have shown significant hematological responses to interferon-α, with improved hemoglobin levels and decreased dyserythropoiesis.4,8
For the treatment of the iron overload, iron chelators are the only option. The chelation treatment in CDA patients generally follows the guidelines for chelation in thalassemia.89 Therefore, early diagnosis of these conditions is critical to prevent iron overload. It is important to periodically evaluate the iron balance parameters in the serum, such as ferritin, transferrin saturation, and hepcidin, and to further evaluate liver and heart iron accumulation using a noninvasive technique, such as T2*-weighted magnetic resonance imaging.
Splenectomy is another therapeutic option under these conditions. Recent recommendations suggested for CDAII include splenectomy only in severely anemic patients and/or in those with symptomatic splenomegaly because of only moderate increases in hemoglobin concentrations after this surgical intervention.90 Of note, cholecystectomy is not performed at the same time of splenectomy anymore, but only if symptomatic lithiasis or other complications are diagnosed.90
New studies are testing drugs to treat CDAII in the preclinical phase. In particular, activin receptor (ActR) ligand traps (eg, luspatercept, sotatercept) are drugs that target ineffective erythropoiesis, and these have shown encouraging results in phase 1 and 2 clinical trials for patients with β-thalassemia.91 These drugs act as ligand traps against ligands of the transforming growth factor-β superfamily by inhibition of its binding to ActRIIA, and thus suppressing the SMAD2/3 pathway (Figure 4). Because of the shared clinical findings between patients with β-thalassemia and CDAII, an ongoing study is evaluating the use of a murine analog of sotatercept, RAP-011, in a CDAII cellular model. Treatment with RAP-011 suppresses the ActRIIA/B pathway, which in turn increases the nuclear levels of the GATA1 transcription factor, and leads to increased expression of GATA1-activated genes involved in erythroid development.92 These data support the beneficial role of sotatercept also in patients with CDAII for the restoration of erythroid maturation.
Another intriguing field of interest is gene therapy. For instance, a recent study described lentiviral transduction of p60-BBF2H7, a transactivator of Sec23a, in primary human erythroblasts.93 In vitro gene therapy with this lentivirus led to upregulation of SEC23A and subsequent normalization of SEC23 levels to compensate for mutated SEC23B in CDAII patients.66,93
Conclusions
CDAs have puzzled hematologists for a long time. CDAs are characterized by elevated clinical and genetic heterogeneities that result in difficulties in reaching a correct differential diagnosis. To diagnose these conditions, it is crucial to have detailed phenotyping and to perform correct genetic testing. Next-generation sequencing testing allows time-effective diagnosis, as well as identification of polygenic conditions and modifier variants. The increased knowledge of the genetic features and the detailed phenotyping of these patients will facilitate their diagnosis, improve their personalized clinical management, and generate advanced telemedicine tools, as has been proposed recently.94
The morphological appearance of CDA erythroblasts suggests their defective development into mature erythrocytes. Although several CDA-associated genes have been identified, how their altered functions lead to erythroblast multinuclearity is often not clear, although studies of the pathogenic mechanisms of CDAs continue to report new achievements that are useful for the clinical management of these patients, and for the study of new therapeutic strategies. Our hope for the future will be the development of specific drugs for patients with CDAs that are useful against both the anemia and the iron overload. The trap ligands appear to be promising under these conditions, as well as gene therapy.
Acknowledgments
This work was supported by the Italian Ministry of University and Research (RBSI144KXC) (R.R.), Programme STAR, UniNA and Compagnia di San Paolo (R.R.), Spanish foundation “Ramón Areces” (CoDysAn project) (A.I.), and a Junior Research Grant (2018, 3978026) from the European Hematology Association (I.A.).
Authorship
Contribution: A.I., I.A., and R.R. designed the study and prepared the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Achille Iolascon, Department of Molecular Medicine and Medical Biotechnologies, “Federico II” University of Naples, 80145 Naples, Italy; e-mail: achille.iolascon@unina.it.
