

Patients with COVID-19 are susceptible to thrombosis and multiorgan failure. In a prospective study in this issue of Blood, 1 identify neutrophil extracellular traps (NETs) as the potential culprits of COVID-19-related pulmonary dysfunction and death.
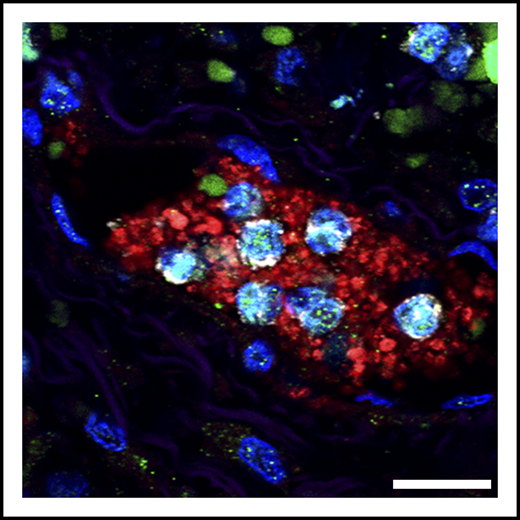
Detail of neutrophils (gray) bearing markers of NETs (citrullinated histone; green), trapped in a platelet-rich clot (red). These microthrombi are found in the lungs of patients with severe COVID-19. Scale bar, 20 μm. See Figure 3A in the article by Middleton et al that begins on page 1169.
Detail of neutrophils (gray) bearing markers of NETs (citrullinated histone; green), trapped in a platelet-rich clot (red). These microthrombi are found in the lungs of patients with severe COVID-19. Scale bar, 20 μm. See Figure 3A in the article by Middleton et al that begins on page 1169.
With >14 million affected individuals and a half million deaths now officially caused by SARS-CoV-2 infection worldwide, this coronavirus has confirmed the predictions of global viral spread into a pandemic with major public health and social implications. Beyond these critical considerations, COVID-19 has also caused clinicians and basic scientists to reexamine our models of immune and inflammatory responses against pathogens. One of these models deals with immune mechanisms that trigger coagulopathy, a common clinical manifestation seen in COVID-19 patients (see figure).2 Coagulopathy is a hypercoagulable state that can inflict irreversible damage to the lungs and other organs. At the same time, an additional hallmark seen in severe cases of COVID-19 is the presence of elevated neutrophil counts,3 a feature that is shared with other forms of cardiovascular disease.
How are coagulopathy and increased neutrophils related in the context of COVID-19? By searching for potential pathogenic mechanisms, Middleton et al report in this issue of Blood that neutrophils from COVID-19 patients are more prone to release NETs than neutrophils from healthy individuals, or from patients displaying milder forms of the disease. NETs are multimolecular, DNA-based complexes released by neutrophils that allow containment and killing of bacteria and fungi and can also trap and deactivate virus.4 Unfortunately, NETs are also thrombogenic because they contain highly cationic proteins.5 Thus, the particular NET-forming propensity of neutrophils in some COVID-19 patients may be causally associated with the development of acute and severe coagulopathies, even though no such patients were identified in the cohort from this study. More generally, the current findings suggest that the SARS-CoV-2 virus elicits potent immunothrombosis, a protective process that allows the containment of pathogens. Both the presence of thrombi and nuclear and granular proteins contained within the DNA lattice that form NETs create a cytotoxic milieu when neutrophils are recruited en mass, thereby compromising epithelial and vascular integrity and contributing to rapid pulmonary dysfunction. Consistent with this notion, Middleton et al find that the presence in blood of NET byproducts, such as DNA free or associated with myeloperoxidase, correlated well with parameters of lung damage in the patients.
Contrasting with these findings, however, the authors found no correlation between NETs and markers of endothelial damage or active thrombosis, such as D-dimers or von Willebrand factor. This suggests that the NET-thrombosis connection may be more complex than anticipated, possibly because activated platelets and factors of the coagulation cascade interact in undefined ways (by interfering or promoting) with the formation of NETs. Alternatively, the NET-thrombosis connection may be obscured because quantification of NETs in plasma is imprecise, for example, because NETs deposited in affected organs may be no longer detectable or because they are being actively degraded. This limitation raises caution regarding the use of absolute NET levels as a prognostic score for COVID-19 patients.
Among the several features reported here, the authors note that neutrophils from severe COVID-19 cases are more granular. Beyond denoting possible activation of these leukocytes, this finding is more likely to associate with the presence of immature granulocytic cells in the circulation of severe COVID-19 patients. This may be important because mobilization of immature neutrophils from the bone marrow, the organ in which granules are synthesized, implies higher content of cytotoxic compounds or NET-inducing enzymes in these cells and further provides links with studies demonstrating a positive correlation between disease severity and the presence of “developing” neutrophils in the circulation of COVID-19 patients.6 Consistently, studies in mice have demonstrated that higher granule content correlates with the ability of neutrophils to form NETs in models of acute pulmonary inflammation, and that hypergranular neutrophils are those recently released from the marrow.7 These findings, however, contrast with the observation in this and a previous report that plasma from severe COVID-19 patients contains factors that render neutrophils prone to form NETs, suggesting that both cell-intrinsic and environmental changes can incite neutrophil-mediated injury (this study and Zuo et al8 ).
Finally, the present study shows that NET formation by neutrophils from severe COVID-19 patients can be effectively blocked by an endogenous inhibitory peptide ex vivo, raising the possibility of using NET-inhibitory or degrading compounds to protect patients from the most severe forms of COVID-19. Although this approach is indeed appealing, and these authors and others have previously discussed various approaches to target NETs to blunt lung injury in COVID-19 patients,9 it remains critically important to provide causal data to support the involvement of NETs in the pathology of COVID-19. This has been hampered so far by the challenge of developing appropriate animal models (eg, animals expressing human ACE2 at the correct locations), and by the strict biosafety conditions needed for manipulation of the virus. Furthermore, larger cohorts of patients will be needed for the prospective or retrospective determination of NET levels in plasma and other tissues, the presence of distinct neutrophil subsets, or NET-inducing factors in order to substantiate the conclusions made here.
Nonetheless, this study represents a much needed effort if we are to identify new strategies to efficiently protect patients from the devastating consequences of SARS-CoV-2 infection.
Conflict-of-interest disclosure: The author declares no competing financial interests.
