

TO THE EDITOR:
Light chain (AL) amyloidosis is caused by tissue deposition of a monoclonal immunoglobulin light chain in the form of amyloid fibrils.1 In AL amyloidosis, amyloid deposition can be systemic or localized.2 Localized amyloidosis is usually a benign disease, although local progression is possible.3 Differently, systemic AL amyloidosis is a severe disease, characterized by progressive vital organ dysfunction that is fatal if not treated timely and effectively.4 In the vast majority of cases, systemic AL amyloidosis is caused by an underlying plasma cell clone that genetically resembles monoclonal gammopathy of undetermined significance (MGUS) or multiple myeloma.5-7 In 5% to 7% of patients, systemic AL amyloidosis is associated with an immunoglobulin M (IgM)-producing clone, with characteristics of MGUS or Waldenström macroglobulinemia, which harbor the MYD88L265P mutation in almost three-fourths of cases.8,9
It is well recognized that IgM-related systemic AL amyloidosis is a distinct clinical entity with peculiar organ involvement and specific prognostic determinants.10-16 Rarely, systemic and localized AL amyloidosis can be associated with nonlymphoplasmacytic lymphoproliferative disorders (LPDs), requiring a distinct, sometimes challenging diagnostic and therapeutic approach. Differently from IgM–AL amyloidosis, this rare association has not yet been systematically studied. Only a few, small case series reported an association between marginal zone lymphoma (MZL) of mucosa-associated lymphoid tissue and localized AL amyloidosis, mainly with nodular pulmonary involvement.17-19 Recently, German investigators published a series of 29 patients with AL amyloidosis and localized B-cell neoplasia, mostly with an MZL immunophenotype20 ; only 5 patients had systemic lymphoma, and 2 were classified as systemic AL amyloidosis.
In the present study, we report the clinical presentation and outcome of 36 patients with nonlymphoplasmacytic LPD and AL amyloidosis.
The prospectively maintained database of the Pavia Amyloidosis Research and Treatment Center, including 1415 treatment-naive patients with AL amyloidosis diagnosed between 2004 and 2015 (1065 systemic and 350 localized), was searched for patients with a diagnosis of nonlymphoplasmacytic lymphoma. Patients with a concomitant bone marrow plasma cell infiltrate consistent with a diagnosis of MGUS or multiple myeloma were excluded from the study, as were those with nonconclusive results of bone marrow studies. The amyloid deposits were characterized as AL-type according to immunoelectron microscopy or mass spectrometry in all cases.21,22 Localized amyloidosis was defined as nodular AL-type amyloid deposition (amyloidoma) in the absence of organ dysfunction.2 The patients with systemic amyloidosis were stratified according to the current cardiac and renal staging systems based on biomarkers.23-25 All patients gave written informed consent for their clinical data to be used in retrospective studies in accordance with the Declaration of Helsinki.
Survival curves were plotted according to the Kaplan-Meier method, and differences in survival were tested for significance with the log-rank test. MedCalc Statistical Software version 14.12.0 (MedCalc Software bvba, Ostend, Belgium; http://www.medcalc.org; 2014) was used for computation.
A total of 36 patients with AL amyloidosis and a nonlymphoplasmacytic LPD were identified. Twenty-one (58%) patients had systemic AL amyloidosis, and 15 (42%) had localized AL amyloidosis. They represented 2% and 5% of all patients with systemic and localized AL amyloidosis referred to our center in the study period, respectively. Patient characteristics are reported in Table 1. Nineteen (53%) patients were diagnosed with MZL, and 11 (58%) of these cases were extranodal.
Characteristics of 36 patients with AL amyloidosis and nonlymphoplasmacytic LPDs
| Characteristic | Systemic AL amyloidosis, 21 patients (No. [IQR or %]) | Localized AL amyloidosis, 15 patients (No. [IQR or %]) |
|---|---|---|
| Age, y | 63 (63-69) | 70 (64-74) |
| Male sex | 15 (71) | 9 (60) |
| MZL | 9 (43) | 10 (67) |
| Extranodal/nodal/disseminated/NOS | 4 (19)/0/1 (5)/4 (19) | 7 (46)/1 (7)/1 (7)/1 (7) |
| Non-MZL | 12 (57) | 5 (33) |
| Low-grade B-cell lymphoma NOS | 5 (24) | 2 (13) |
| DLBCL/CLL-SLL/other diagnosis* | 2 (10)/2 (10)/3 (13) | 2 (13)/1 (7)/0 |
| Organ involvement in systemic AL amyloidosis | NA | |
| Heart/kidney/liver/soft tissues/ANS/PNS | 12 (57)/8 (38)/1 (5)/4 (19)/3 (13)/3 (13) | |
| Site of localized AL amyloidosis | NA | |
| Nodular pulmonary/lymph nodes | 7 (46)/3 (10) | |
| Skin/tracheobronchial/bladder/nodular GI | 2 (13)/1 (7)/1 (7)/1 (7) | |
| NT-proBNP, ng/L | 1113 (558-6979) | 152.28 (47-265) |
| Proteinuria, g/24 h | 1.21 (0.10-5.00) | 0.07 (0.06-0.09) |
| Cardiac stage I/II/IIIa/IIIb† | 8 (32)/11 (44)/4 (16)/2 (8) | NA |
| Renal stage I/II/III‡ | 15 (60)/8 (32)/2 (8) | NA |
| Ann Arbor stage I/II/III/IV | 2 (10)/1 (5)/1 (5)/17 (80) | 8 (53)/0/2 (13)/5 (34) |
| Monoclonal component at serum and/or urine immunofixation | 20 (95) | 8 (53) |
| Kappa: lambda ratio | 4 (20): 16 (80) | 4 (50): 4 (50) |
| Monoclonal component | ||
| IgGλ/IgGκ/IgAλ/IgAκ | 1 (5)/6 (28)/1 (5)/1 (5) | 1 (7)/3 (20)/0/0 |
| IgMλ/IgMκ/FLC λ/FLC κ | 2 (10)/2 (10)/5 (24)/0 | 2 (13)/0/1 (7)/1 (7) |
| Free light chain only: complete monoclonal component | 6 (29): 14 (67) | 2 (25): 6 (75) |
| Positive fat pad aspirate (Congo red) | 17 (80) | 0 |
| Abnormal FLCR | 15 (71) | 5 (33) |
| B symptoms | 3 (13) | 1 (7) |
| HCV/HBV infection | 2 (10)/3 (13) | 0/3 (20) |
| Treatment of lymphoma | 14 (66) | 10 (67) |
| R-CHOP/R-CVP/radiotherapy | 7 (33)/2 (10)/0 | 5 (34)/1 (7)/2 (13) |
| Alkylating agents/other treatment§ | 3 (13)/2 (10) | 0/2 (13) |
| Characteristic | Systemic AL amyloidosis, 21 patients (No. [IQR or %]) | Localized AL amyloidosis, 15 patients (No. [IQR or %]) |
|---|---|---|
| Age, y | 63 (63-69) | 70 (64-74) |
| Male sex | 15 (71) | 9 (60) |
| MZL | 9 (43) | 10 (67) |
| Extranodal/nodal/disseminated/NOS | 4 (19)/0/1 (5)/4 (19) | 7 (46)/1 (7)/1 (7)/1 (7) |
| Non-MZL | 12 (57) | 5 (33) |
| Low-grade B-cell lymphoma NOS | 5 (24) | 2 (13) |
| DLBCL/CLL-SLL/other diagnosis* | 2 (10)/2 (10)/3 (13) | 2 (13)/1 (7)/0 |
| Organ involvement in systemic AL amyloidosis | NA | |
| Heart/kidney/liver/soft tissues/ANS/PNS | 12 (57)/8 (38)/1 (5)/4 (19)/3 (13)/3 (13) | |
| Site of localized AL amyloidosis | NA | |
| Nodular pulmonary/lymph nodes | 7 (46)/3 (10) | |
| Skin/tracheobronchial/bladder/nodular GI | 2 (13)/1 (7)/1 (7)/1 (7) | |
| NT-proBNP, ng/L | 1113 (558-6979) | 152.28 (47-265) |
| Proteinuria, g/24 h | 1.21 (0.10-5.00) | 0.07 (0.06-0.09) |
| Cardiac stage I/II/IIIa/IIIb† | 8 (32)/11 (44)/4 (16)/2 (8) | NA |
| Renal stage I/II/III‡ | 15 (60)/8 (32)/2 (8) | NA |
| Ann Arbor stage I/II/III/IV | 2 (10)/1 (5)/1 (5)/17 (80) | 8 (53)/0/2 (13)/5 (34) |
| Monoclonal component at serum and/or urine immunofixation | 20 (95) | 8 (53) |
| Kappa: lambda ratio | 4 (20): 16 (80) | 4 (50): 4 (50) |
| Monoclonal component | ||
| IgGλ/IgGκ/IgAλ/IgAκ | 1 (5)/6 (28)/1 (5)/1 (5) | 1 (7)/3 (20)/0/0 |
| IgMλ/IgMκ/FLC λ/FLC κ | 2 (10)/2 (10)/5 (24)/0 | 2 (13)/0/1 (7)/1 (7) |
| Free light chain only: complete monoclonal component | 6 (29): 14 (67) | 2 (25): 6 (75) |
| Positive fat pad aspirate (Congo red) | 17 (80) | 0 |
| Abnormal FLCR | 15 (71) | 5 (33) |
| B symptoms | 3 (13) | 1 (7) |
| HCV/HBV infection | 2 (10)/3 (13) | 0/3 (20) |
| Treatment of lymphoma | 14 (66) | 10 (67) |
| R-CHOP/R-CVP/radiotherapy | 7 (33)/2 (10)/0 | 5 (34)/1 (7)/2 (13) |
| Alkylating agents/other treatment§ | 3 (13)/2 (10) | 0/2 (13) |
Variables from patients with systemic and localized AL amyloidosis were tested for statistical significance if applicable. ANS, autonomic nervous system; CLL-SLL, chronic lymphocytic leukemia–small lymphocytic lymphoma; DLBCL, diffuse large B-cell lymphoma; FLCR, free light chain ratio; HBV, hepatitis B virus; HCV, hepatitis C virus; GI, gastrointestinal; NA, not applicable; NOS, not otherwise specified; NT-proBNP, N-terminal pro–B-type natriuretic peptide; PNS, peripheral nervous system; R-CHOP, rituximab, cyclophosphamide, adriamycin, vincristine, prednisone; R-CVP, rituximab, cyclophosphamide, vincristine, prednisone.
Other diagnosis: 1 follicular lymphoma, 1 hairy cell leukemia, and 1 Hodgkin lymphoma.
Cardiac stage: Based on NT-proBNP (cutoff, 332 ng/L) and cardiac troponin I (cutoff, 0.1 ng/mL). Stage I patients have both markers below the cutoffs; stage II patients, only one marker above the cutoffs; and stage III patients, both markers above the cutoffs. NT-proBNP >8500 ng/L divided stage III patients into stage IIIa and IIIb cases.
Renal stage: Stage I, both proteinuria ≤5 g/24 hours and estimated glomerular filtration rate (eGFR) ≥50 mL/min per 1.73 m2; stage II, either proteinuria >5 g/24 hours or eGFR <50 mL/min per 1.73 m2; stage III, both proteinuria >5 g/24 hours and eGFR <50 mL/min per 1.73 m2.
Other treatment: 1 fludarabine and cyclophosphamide and rituximab, and 1 cladribine in patients with systemic AL amyloidosis and 1 rituximab in monotherapy, 1 chlorambucil in patients with localized AL amyloidosis
Autoimmune disorders were more frequent in patients with localized amyloidosis (53% vs 5%; P = .001). Sjögren syndrome was the most common autoimmune disease (6 of 9 patients). In the overall cohort, patients with systemic amyloidosis were more likely to have advanced Ann Arbor stage than subjects with localized amyloidosis (85% vs 46%, respectively; P = .006). A serum and/or urinary monoclonal component and/or an abnormal free light chain ratio were present in all patients with systemic AL amyloidosis and in 6 subjects (54%) with localized amyloid deposits (P = .002).
In 14 of 21 patients with systemic AL amyloidosis, the diagnosis of lymphoma preceded the clinical manifestations of amyloidosis by a median of 32 months (interquartile range [IQR], 7-74 months). The diagnosis of systemic amyloidosis was established after a median of 64 months (IQR, 40-84 months) from the diagnosis of lymphoma, with a median diagnostic delay of 24 months (IQR, 11-33 months) from the onset of symptoms of amyloidosis, despite 16 (64%) of these subjects having a monoclonal component detected at the time of the lymphoma diagnosis. In the remaining 7 cases, lymphoma was diagnosed during the investigations for systemic amyloidosis.
Overall, 24 (64%) patients received therapy for LPD, before the diagnosis of AL amyloidosis. Sixteen (44%) were treated with a rituximab-based regimen, and after a median of 5 cycles (range, 2-8 cycles), 17 (74%) subjects achieved a response (complete in 13 [57%]) for lymphoma. Treatment of systemic amyloidosis was bortezomib based in 11 patients, rituximab based in 7, and oral melphalan-dexamethasone based in 3. Eleven patients (52%) achieved a hematologic response that was complete in 2 subjects and very good partial response in 6. The median follow-up of living patients was 16 months. Overall, 12 patients with systemic amyloidosis died, 11 due to progression of amyloidosis and 1 because of an unrelated cause (gastric cancer). None of the patients with systemic amyloidosis died of progressive lymphoma. The median survival from the diagnosis of systemic amyloidosis was 26 months (Figure 1).
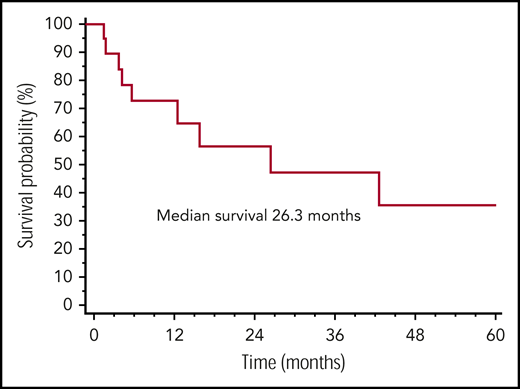
Overall survival in systemic AL amyloidosis and nonlymphoplasmacytic LPDs. Overall survival from diagnosis of AL amyloidosis of 21 patients with systemic amyloidosis and nonlymphoplasmacytic LPDs.
Overall survival in systemic AL amyloidosis and nonlymphoplasmacytic LPDs. Overall survival from diagnosis of AL amyloidosis of 21 patients with systemic amyloidosis and nonlymphoplasmacytic LPDs.
To the best of our knowledge, this is the largest clinical series of patients with AL amyloidosis and nonlymphoplasmacytic LPD published thus far. We observed that MZL is the most common nonlymphoplasmacytic LPD associated with AL amyloidosis, but other types of LPD can also underlie light chain amyloid deposition. It seems that more advanced-stage lymphomas were more likely to give rise to systemic amyloidosis. Conversely, autoimmune disorders, mostly Sjögren syndrome, seemed to be associated with localized disease.
Systemic amyloidosis portended a poor outcome, being the leading cause of death in our series. Importantly, we observed a significant diagnostic delay in this group of patients, even in those followed up by hematologists for the underlying lymphoma. Earlier diagnosis could improve the outcome of these patients, and the presence of a monoclonal component or an abnormal free light chain ratio can be clues to the diagnosis of systemic AL amyloidosis in patients with LPD. Fat aspiration is a valuable diagnostic tool, as it was positive in the 80% of patients with systemic amyloidosis. However, organ biopsy may be necessary in patients with a strong clinical suspicion even if the abdominal fat aspirate is negative.
Hematologists should be aware that systemic amyloidosis can be a life-threatening complication of nonlymphoplasmacytic LPD. Large international studies are warranted to better characterize this rare association and to identify appropriate therapeutic interventions.
For original data, please contact giovanni.palladini@unipv.it.
This study was presented in abstract form at the 60th annual meeting of the American Society of Hematology, San Diego, CA, 1 December 2018.
Acknowledgments
This work was supported by Fondazione Cariplo (grant nos. 2014-0700, 2015-0591, and 2016-0489), Associazione Italiana per la Ricerca sul Cancro special program “5 per mille” (no. 9965), the Italian Ministry of Health (grants RF-2013-02355259 and RF-2016-02361756), the Italian Medicines Agency (grant AIFA-2016-02364602), and E-Rare JTC 2016 grant ReDox. G.P. is supported in part by the Bart Barlogie Young Investigator Award from the International Myeloma Society. P.M. is supported in part by the Fellowship award from Ghislieri College. M.N. is supported in part by a grant from the Amyloidosis Foundation. This research was funded by the Italian Ministry of Education, University and Research to the Departments of Molecular Medicine and Biology and Biotechnologies of the University of Pavia under the initiative “Dipartimenti di Eccellenza (2018-2022).”
Authorship
Contribution: L.A. and G.P. designed the study; M.B., I.D., L.A., and G.P. evaluated patients, collected data, analyzed data, wrote the manuscript, and gave final approval; P.M., M.N., A.F., and G.M. evaluated patients, critically reviewed the manuscript, and gave final approval; and S.R., S.M., M.V., P.B., C.S.C., and M.P. critically reviewed the manuscript and gave final approval.
Conflict-of-interest disclosure: G.P. receives honoraria from Janssen, honoraria and travel support from Prothena, and travel support from Celgene. M.N. receives honoraria from Janssen. L.A. reports consulting or advisory roles for Bayer, Celgene, Gilead Sciences, Roche, Sandoz, Janssen-Cilag, and Verastem; receives research funding from Gilead Sciences; participates in a speakers bureau for Celgene; and receives travel expenses from Celgene, Roche, Janssen-Cilag, and Gilead. G.M. is a consultant for Millennium Pharmaceuticals Inc., Pfizer, Janssen, Prothena, and IONIS. The remaining authors declare no competing financial interests.
Correspondence: Giovanni Palladini, Amyloidosis Research and Treatment Center, Fondazione IRCCS Policlinico San Matteo, Viale Golgi, 19-27100 Pavia, Italy; e-mail: giovanni.palladini@unipv.it; or Luca Arcaini, Division of Hematology, Fondazione IRCCS Policlinico San Matteo, Viale Golgi, 19-27100 Pavia, Italy; e-mail: luca.arcaini@unipv.it.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal