

Key Points
A genetic predisposition to CH is not detectable in this cohort of 594 elderly twins.
CHIP mutations are not associated with mortality in a direct comparison among discordant twin pairs.

Abstract
Clonal hematopoiesis (CH) of indeterminate potential (CHIP) is defined by mutations in myeloid cancer–associated genes with a variant allele frequency of at least 2%. Recent studies have suggested a possible genetic predisposition to CH. To further explore this phenomenon, we conducted a population-based study of 594 twins from 299 pairs aged 73 to 94 years, all with >20 years’ follow-up. We sequenced DNA from peripheral blood with a customized 21-gene panel at a median coverage of 6179X. The casewise concordance rates for mutations were calculated to assess genetic predisposition. Mutations were identified in 214 (36%) of the twins. Whereas 20 twin pairs had mutations within the same genes, the exact same mutation was only observed in 2 twin pairs. No significant difference in casewise concordance between monozygotic and dizygotic twins was found for any specific gene, subgroup, or CHIP mutations overall, and no significant heritability could be detected. In pairs discordant for CHIP mutations, we tested if the affected twin died before the unaffected twin, as a direct measurement of the association of having CH when controlling for familial factors. A total of 127 twin pairs were discordant for carrying a mutation, and in 61 (48%) cases, the affected twin died first (P = .72). Overall, we did not find a genetic predisposition to CHIP mutations in this twin study. The previously described negative association of CHIP mutations on survival could not be confirmed in a direct comparison among twin pairs that were discordant for CHIP mutations.
Introduction
More than 20 years ago, studies of X-chromosome inactivation in elderly women suggested that selected hematopoietic clones expand in the aging bone marrow.1 Our studies of aging twins proposed that this aberrant hematopoietic stem cell kinetics is not random.2 The accumulation of clonal stem cells has previously been associated with myeloid skewing, lymphopenia, and progressing anemia in the elderly.3
In recent years, comprehensive exome and targeted sequencing studies of large cohorts revealed that the hematopoietic stem cells of healthy elderly individuals accumulate somatic mutations in the same genes that are drivers of myeloid cancer, thus giving rise to clonal hematopoiesis (CH).4-6 Subsequent studies have confirmed and extended these findings,7-10 and several terms have been proposed to describe the condition, including age-related clonal hematopoiesis, which signifies CH of any acquired clonal event,1,11 and clonal hematopoiesis of indeterminate potential (CHIP), which has been defined to require a variant allele frequency (VAF) of driver genes of a minimum of 2%.9 Ultradeep sequencing studies have revealed that very small clones may be ubiquitously present at a young age12 ; however, the prevalence of CHIP increases dramatically after middle age.4,5,8,10
The most frequent aberrations in CHIP are loss-of-function mutations in the epigenetic modifiers DNMT3A, TET2, and ASXL1, so-called DTA mutations. Stem cells with mutations in DNMT3A had already been shown to precede the development of acute myeloid leukemia (AML),13 when analyses of the large cohorts revealed that CHIP carriers have a significantly increased (10-fold) risk of hematological cancers, including, but not restricted to, AML and myelodysplastic syndrome (MDS).4,5 This was particularly the case in patients with CHIP mutations of a VAF > 10%, where the risk of hematological cancer was increased by a factor of 50.5 Further studies have shown that CHIP mutations frequently persist in patients in complete remission after treatment of AML,14,15 but only the non-DTA mutations are associated with an increased risk of relapse.15
Twin studies from Scandinavia have shown an increased familial risk of cancer, and in chronic myeloid neoplasms, Andersen et al16 demonstrated a casewise concordance (cc) rate of 15% among monozygotic (MZ) twins compared with 0% among dizygotic (DZ) twins (P = .016), suggesting a genetic predisposition. In addition, it has been known for decades that some germline aberrancies predispose to hematological cancer, such as AML and MDS. In some of these genes, driver mutations occur both in the germline and as somatic events (eg, in RUNX1, GATA2, and ETV6). When patients with predisposing germline variants are diagnosed with overt AML or MDS, cooccurring somatic mutations, which are involved in leukemogenesis, are often present.17
In adults, hematological cancers are rarely caused by germline aberrancies, although recent sequencing studies have identified germline mutations that predispose to hereditary myeloid cancers after middle age.18 Although the link between somatic CHIP mutations and hematological cancer is evident, it is unknown why some aging individuals develop CHIP while others do not. One Icelandic study suggested that besides age, smoking and psychiatric disease are associated with CH. They performed a genome-wide association study and identified an 8 base-pair (bp) deletion in TERT intron 3 (RS 34002450)19 with an increased risk of CH with an odds ratio of 1.37 and an allele frequency of 40.6% in Iceland. Meanwhile, Buscarlet et al described a genetic predisposition to TET2 mutations within sibships with a recurrence risk of 0.09 and 0.13 corresponding to a recurrence risk ratio of 2.2 and 2.7 in age groups >54 and 64, respectively. However, no association was observed for DNMT3A mutations.20 Last, Frick et al recently investigated CHIP in related donors to patients undergoing allogeneic stem cell transplantation. They identified a higher prevalence of CHIP in related donors to patients with a myeloid malignancy compared with a lymphoid malignancy of 19.2% vs 6.3% (P < .001), also suggesting a possible familial predisposition.21
However, these studies were not designed to distinguish whether CHIP is resulting from a common germline predisposition, and whether such an aberration also predisposes to hematological cancer. In addition, it is not clear whether the reported aggregation of TET2 mutations in elderly siblings is merely driven by a genetic predisposition or by common environmental factors. To clarify whether CHIP mutations arise on a common genetic basis or are a result of shared environmental exposures, we conducted a study of 299 elderly twin pairs who were prospectively followed for >20 years.
Methods
Cohort
In the present study, twin pairs were identified from the Longitudinal Study of Aging Danish Twins (LSADT) in which 689 twins donated blood in 1997,22 and 299 intact same-sex twin pairs were included in the present study. Twin zygosity was classified using questionnaire-based zygosity assessment, which is found to be correct in 95% of the cases.23 Information about smoking (pack-years) was obtained from the LSADT questionnaire in 1997. The LSADT study was approved by the Regional Committees on Health Research Ethics for Southern Denmark (S-VF-20040241) and the present study (S-20170053). Research was conducted in accordance with the Declaration of Helsinki.
Sequencing and bioinformatic analyses
A targeted sequencing approach was applied, using an Illumina TruSeq Custom Amplicon panel (Illumina, San Diego, CA) covering >95% of mutations commonly associated with CH4-6,24 (supplemental Material, available on the Blood Web site). DNA was quantified using the Qubit fluorometer (Life Technologies, Carlsbad, CA), and 100 to 200 ng of DNA was used for library preparation. To optimize variant calling and identification of low-level mutations, unique molecular identifiers, consisting of 6 random index nucleotides, were added to each sample before amplification. Libraries were pooled and sequenced on the Illumina NextSeq platform (Illumina). We used a 300-cycle mid output kit as specified by the manufacturer.
Variant calling
In brief, raw sequence reads and corresponding unique molecular identifiers were aligned to the human reference genome (hg19 build) with the BWA-mem algorithm.25 Identification of possible variants was performed using 2 variant callers: Freebayes v.1.1.026 and VarDict v.1.5.1.27 We investigated CHIP mutations, as defined by Steensma et al,9 with a VAF of 2% or above. Common variants, defined by a population frequency of >1% in large public variant databases (ExAC, TOPMED, 1000Genomes), were excluded, and furthermore, we removed rare variants with a VAF of 40% to 60%, which were concordant in related twins, so these should not bias the cc rates. Additional details are provided in the supplemental Material.
Vital status and diagnoses
Date of birth, vital status, and date of death were retrieved from the Danish Civil Registration System.28 To investigate the role of CHIP mutations for the risk of subsequent hematological cancer or cytopenia, we extracted diagnoses from The Danish National Patient Register, which essentially contains all discharges from Danish hospitals since 1977.29 From 1994 and onward, diagnoses were classified according to the International Classification of Diseases, Tenth Revision. In the present study, we included primary and secondary diagnoses and all patient types (inpatient, outpatient, emergency room patient) from 1997 to 2014. We defined 2 groups of International Classification of Diseases, Tenth Revision codes representing hematological cancers or cytopenia, respectively (supplemental Materials).
Statistical analyses
Concordance and heritability
The classic twin-study methodology is based on MZ twins having identical genotypes, whereas DZ twins share, on average, half of their genetic variants like biologic full siblings. A greater phenotypic similarity in MZ than in DZ twins is expected if there is a substantial genetic component in the etiology of the phenotype. We assessed the similarity of MZ and DZ twins using cc rates and tetrachoric correlations. The cc rate is defined as the probability that a twin is affected given the cotwin is affected and corresponds to recurrence rates in siblings. Assuming that there is a normally distributed liability to develop a complex phenotype, we used standard quantitative biometric models to estimate the tetrachoric correlations and the relative contribution of genetic and environmental factors in CHIP occurrence30
ADCE model
The phenotypic variance can then be separated into 4 variance components: variance attributable to additive genetic effects (A), genetic dominance (D), shared environment (C), and nonshared (individual-specific) environment (E). Only nonshared environments contribute to dissimilarity within MZ twin pairs because of their presumed genetic identity, whereas the effects of additive genetic factors and genetic dominance may also contribute to dissimilarity within DZ pairs, who share, on average, half of the additive and one-quarter of the dominant genetic factors.30
Time to event analyses
Follow-up was initiated at the date of blood donation. Cox proportional hazard model with time since blood sample was used as the underlying time scale to compute hazard ratios (HRs) of time to death for individuals with CHIP mutations compared with individuals without CHIP mutations. In these analyses, the follow-up was terminated at the date of death or 1 August 2018, whichever came first.
To assess the risk of developing hematological cancer or cytopenia, competing risks regression analyses were performed treating death as a competing risk using the STATA command stcrreg, which is based on Fine and Gray’s proportional subhazards model. Follow-up was terminated at date of first diagnosis of the specific diagnosis group, death, or 1 March 2014, whichever came first.
In addition, because some studies indicate that bias can be introduced using time since sampling as a time scale,31,32 all time-to-event analyses were also performed with age as the underlying time scale.
The proportional hazard assumption was assessed based on Schoenfeld residuals. If the proportional hazard assumption was violated for the covariates, we performed Cox regression with strata on covariates to assess if changes in results are noteworthy.
Furthermore, in twins discordant for mutation, we tested if the affected twin died before the unaffected twin using the binomial probability test.
In addition, all analyses were performed for different classifications of mutations (DNMT3A, TET2, and DTA mutations overall) and based on VAF.
Results
In this study, we included a total of 598 elderly twins from 299 pairs, which were followed prospectively for 20 years. Four twins were excluded from the study due to poor sequencing quality, and their corresponding twin was removed from the concordance analyses, which then included a total of 295 twin pairs. The median age at inclusion was 77 years (range 73 to 94); 33% of the twins were male twins, whereas 256 (43%) and 338 (57%) were MZ and DZ twins, respectively (Table 1). By targeted next-generation sequencing of DNA from peripheral blood drawn at study entry, we identified a total of 286 mutations in 15 known, recurrent CHIP genes, distributed among 214 (36%) twins (Figures 1 and 2). Thus, some twins carried >1 mutation, with a maximum of 3 mutations being detected in 1 individual (supplemental Figure 1).
Clinical characteristics
| CHIP-mutation negative | CHIP-mutation positive | Total | |||||
|---|---|---|---|---|---|---|---|
| N | % | N | % | P | N | % | |
| Total | 380 | 63.97 | 214 | 36.03 | 594 | 100.00 | |
| Men | 127 | 33.42 | 69 | 32.24 | .769 | 196 | 33.00 |
| Age at blood collection, y | |||||||
| 73-75 | 72 | 18.95 | 29 | 13.55 | <.001 | 101 | 17.00 |
| 75-80 | 205 | 53.95 | 101 | 47.20 | 306 | 51.52 | |
| 80-85 | 82 | 21.58 | 47 | 21.96 | 129 | 21.72 | |
| 85-93 | 21 | 5.53 | 37 | 17.29 | 58 | 9.76 | |
| Zygosity | |||||||
| MZ | 155 | 40.79 | 101 | 47.20 | .130 | 256 | 43.10 |
| DZ | 225 | 59.21 | 113 | 52.80 | 338 | 56.90 | |
| Tobacco consumption (pack-years) | |||||||
| 0 | 146 | 38.42 | 79 | 36.92 | .815 | 225 | 37.88 |
| 1st tertile (1-8.55) | 7 | 20.00 | 40 | 18.69 | 116 | 19.53 | |
| 2nd tertile (8.55-25) | 77 | 20.26 | 46 | 21.50 | 123 | 20.71 | |
| 3rd tertile (25+) | 65 | 17.11 | 43 | 20.09 | 108 | 18.18 | |
| Missing | 16 | 4.21 | 6 | 2.80 | 22 | 3.70 | |
| Dead prior to August 2018 | 359 | 94.47 | 210 | 98.13 | .033 | 569 | 95.79 |
| CHIP-mutation negative | CHIP-mutation positive | Total | |||||
|---|---|---|---|---|---|---|---|
| N | % | N | % | P | N | % | |
| Total | 380 | 63.97 | 214 | 36.03 | 594 | 100.00 | |
| Men | 127 | 33.42 | 69 | 32.24 | .769 | 196 | 33.00 |
| Age at blood collection, y | |||||||
| 73-75 | 72 | 18.95 | 29 | 13.55 | <.001 | 101 | 17.00 |
| 75-80 | 205 | 53.95 | 101 | 47.20 | 306 | 51.52 | |
| 80-85 | 82 | 21.58 | 47 | 21.96 | 129 | 21.72 | |
| 85-93 | 21 | 5.53 | 37 | 17.29 | 58 | 9.76 | |
| Zygosity | |||||||
| MZ | 155 | 40.79 | 101 | 47.20 | .130 | 256 | 43.10 |
| DZ | 225 | 59.21 | 113 | 52.80 | 338 | 56.90 | |
| Tobacco consumption (pack-years) | |||||||
| 0 | 146 | 38.42 | 79 | 36.92 | .815 | 225 | 37.88 |
| 1st tertile (1-8.55) | 7 | 20.00 | 40 | 18.69 | 116 | 19.53 | |
| 2nd tertile (8.55-25) | 77 | 20.26 | 46 | 21.50 | 123 | 20.71 | |
| 3rd tertile (25+) | 65 | 17.11 | 43 | 20.09 | 108 | 18.18 | |
| Missing | 16 | 4.21 | 6 | 2.80 | 22 | 3.70 | |
| Dead prior to August 2018 | 359 | 94.47 | 210 | 98.13 | .033 | 569 | 95.79 |
We included 299 twin pairs and sequenced peripheral blood DNA from all twins. Four twins had low-quality DNA and were excluded from the study, and 594 remaining twins were included in the study. In August 2018, 96% of the cohort had died, and follow-up was available for all twins. Only 38% of the cohort never smoked; the smokers were divided into tertiles according to tobacco consumption for survival analysis.
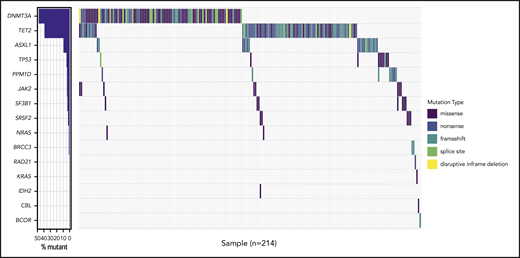
Waterfall plot of the 214 twins with a total of 286 mutations. Waterfall plot of the 214 (36%) cases with at least 1 mutation detected, accounting for a total of 286 mutations being 114 missenses, 76 frameshifts, 69 nonsenses, 21 splice sites, and 5 inframe deletions.
Waterfall plot of the 214 twins with a total of 286 mutations. Waterfall plot of the 214 (36%) cases with at least 1 mutation detected, accounting for a total of 286 mutations being 114 missenses, 76 frameshifts, 69 nonsenses, 21 splice sites, and 5 inframe deletions.

Clone size depicted as a density plot and by individual mutation. We only reported mutations with an allele frequency of 2% and above, which defines CHIP. The distribution of the allele frequencies is shown by density (A), and the variant of each case is shown (B). The median VAF in the entire cohort was 4.4%.
Clone size depicted as a density plot and by individual mutation. We only reported mutations with an allele frequency of 2% and above, which defines CHIP. The distribution of the allele frequencies is shown by density (A), and the variant of each case is shown (B). The median VAF in the entire cohort was 4.4%.
The proportion of individuals with a CHIP mutation was increasing with age ranging from 29% at the age of 75 years to >64% in the group >85 years (supplemental Figure 2). The most commonly mutated gene was DNMT3A observed in 104 twins, followed by TET2, which was mutated in 81 twins. The higher number of mutations in DNMT3A compared with TET2 (17.2% and 10.1%, respectively) was found in the age group of 73 to 79 years. However, in individuals between 80 and 94 years, this trend was reversed, with TET2 mutations being most abundant (ie, 21.4% mutations in TET2 vs 18.2% in DNMT3A) (supplemental Figure 3a-b). In total, DTA mutations accounted for 79.4% of all the mutations observed in this cohort. We identified 96 TET2 mutations: 36 nonsense, 35 frameshift, 19 missense, 5 splice site mutations, and 1 deletion. None of the missense mutations had a VAF >40%. The 115 mutations in DNMT3A were 61 missense, 20 frameshift, 16 nonsense, and 14 splice site mutations, as well as 4 in-frame deletions. Six of the missense mutations were at the hot-spot position R882. Twins with CHIP mutations were older than the nonmutated twins (P ≤ .001), and CHIP mutations were equally distributed among MZ and DZ twins (Table 1). The median VAF in the entire cohort was 4.4% (range = 2% to 68%), with 2 individuals having a VAF > 50%. Based on VAF of the largest clone found in each twin, they were grouped as having <10% vs 10% and higher. Of the 214 mutated cases, 63 (29%) had a VAF of 10% or above, and the frequency increased with age (supplemental Figure 4).
Cooccurrence of mutations in twin pairs
Whereas 20 twin pairs had mutations within the same genes, the exact same mutations (involving the exact same base exchange) were only observed among 2 twin pairs. One twin pair, which was DZ, had an SRSF2 mutation at position c.284C>A causing P95H, with a VAF of 4.5% and 31%, respectively, without any other cooccurring mutations. The second twin pair, which was MZ, had a 5-bp deletion c.912_916delCTGGT in DNMT3A, giving rise to a frameshift at p.Trp305fs with a VAF of 11.7% and 26.8%, respectively. Both of these twins each had an additional DNMT3A mutation; the first twin had a missense mutation c.2096G>T, causing an amino acid change at p.Gly699Val with a VAF of 7.3%, and the second twin had a 4-bp deletion c.2167_2168insGTAG giving rise to a frameshift p.Leu723fs with a VAF of 2%. The common 5-bp deletion with VAF among the MZ twin pair is intriguing, because it could indicate mosaicism due to a deletion occurring at a very early developmental stage; however, it is beyond the scope of the current study to further analyze this issue.
Concordance and heritability
We estimated the cc rates for carrying a mutation in any of the 21 examined genes and found that the concordance rate for MZ twins was 0.40 (0.32; 0.49) and for DZ was twins 0.40 (0.32; 0.49), ccMZ = ccDZ (P = 1). Because TET2 mutations had previously been reported to cooccur in families, whereas DNMT3A did not,20 we further tested the concordance rates for TET2 and DNMT3A separately. For TET2, we observed a concordance rate for MZ and DZ of 0.20 (0.10; 0.34) and 0.20 (0.10; 0.34), respectively, ccMZ = ccDZ (P = 1). Similar results were obtained for DNMT3A 0.25 (0.12; 0.43) and 0.22 (0.11; 0.40), for MZ and DZ twins, respectively, ccMZ = ccDZ (P = .86). When examining all DTA mutations together, we also found similar concordance rates for MZ and DZ twins; 0.33 (0.24; 0.43) and 0.33 (0.24; 0.43), respectively, ccMZ = ccDZ (P = 1). Last, we examined concordance rates of the groups having maximum VAF < 10% vs the group with no mutation. Again, we did not observe any significant difference between MZ or DZ twins using these cutoffs (Table 2). To evaluate the environmental effects, we applied the polygenic ADCE model and estimated what was governing the presence of CHIP mutations, and subsequently, the presence of TET2 and DNMT3A mutations. The biometrical models revealed that 89% (95% confidence interval [CI]: 71% to 100%) of the variation in the liability to have any CHIP mutations was accounted for by environmental factors not shared by cotwins, whereas 0% (95% CI: not applicable) was accounted for by genetic factors and 11% (95% CI: 0% to 29%) was explained by common environmental factors. For TET2 and DNMT3A, the corresponding numbers were 84% (95% CI: 58% to 100%) and 83% (95% CI: 48% to 100%) for environmental factors not shared by cotwins, whereas the genetic factors and common environmental effects were so small that they could not be reliably estimated with the present sample size. Hence, a genetic predisposition to CH is not detectable in this cohort of elderly twins neither overall nor for specific mutations or subgroups.
Concordance rates according to any mutation, DNMT3A, TET2, “DTA mutations,” and maximum clone size <10%
| MZ | DZ | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Number of pairs | Concordant pairs | Discordant pairs | cc rate | Number of pairs | Concordant pairs | Discordant pairs | cc rate | P for ccMZ = ccDZ | |
| Variable | N | N | N | pc (95% CI) | N | N | N | pc (95% CI) | P |
| Any mutation (yes/no) | 81 | 20 | 61 | 0.40 (0.32; 0.49) | 89 | 23 | 66 | 0.40 (0.32; 0.49) | 1 |
| TET2 mutation (yes/no) | 35 | 4 | 31 | 0.20 (0.10; 0.34) | 38 | 4 | 34 | 0.20 (0.10; 0.34) | 1 |
| DNMT3A mutation (yes/no) | 41 | 6 | 35 | 0.25 (0.12; 0.43) | 50 | 6 | 44 | 0.22 (0.11; 0.40) | .86 |
| “Only TET2/DNMT3A/ASXL1” vs “no mutation” | 65 | 14 | 51 | 0.33 (0.24; 0.43) | 63 | 11 | 52 | 0.33 (0.24; 0.43) | 1 |
| “Max VAF <10%” vs “no mutation” | 57 | 11 | 46 | 0.28 (0.25; 0.32) | 57 | 7 | 50 | 0.28 (0.25; 0.32) | .99 |
| MZ | DZ | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Number of pairs | Concordant pairs | Discordant pairs | cc rate | Number of pairs | Concordant pairs | Discordant pairs | cc rate | P for ccMZ = ccDZ | |
| Variable | N | N | N | pc (95% CI) | N | N | N | pc (95% CI) | P |
| Any mutation (yes/no) | 81 | 20 | 61 | 0.40 (0.32; 0.49) | 89 | 23 | 66 | 0.40 (0.32; 0.49) | 1 |
| TET2 mutation (yes/no) | 35 | 4 | 31 | 0.20 (0.10; 0.34) | 38 | 4 | 34 | 0.20 (0.10; 0.34) | 1 |
| DNMT3A mutation (yes/no) | 41 | 6 | 35 | 0.25 (0.12; 0.43) | 50 | 6 | 44 | 0.22 (0.11; 0.40) | .86 |
| “Only TET2/DNMT3A/ASXL1” vs “no mutation” | 65 | 14 | 51 | 0.33 (0.24; 0.43) | 63 | 11 | 52 | 0.33 (0.24; 0.43) | 1 |
| “Max VAF <10%” vs “no mutation” | 57 | 11 | 46 | 0.28 (0.25; 0.32) | 57 | 7 | 50 | 0.28 (0.25; 0.32) | .99 |
Casewise concordance (cc) rates were calculated to estimate a genetic predisposition. If the cc rates are higher in the MZ twins compared with DZ twins, it indicates a genetic predisposition to the variable. We did not find any difference in the cc rates between MZ or DZ in any of the tested variables.
Mortality
At the end of follow-up (August 1, 2018), 96% of the study population was deceased. In a Cox regression analysis, not controlling for covariates, CHIP mutation carriers had an HR (95% CI) of 1.31 (1.10; 1.56) (P < .001). When adjusted for age, sex, and tobacco consumption in a Cox regression analysis, CHIP was borderline significantly associated with increased mortality (HR 1.17 [0.97; 1.40], P = .096) (Table 3). When we used age as the underlying time scale in a Cox regression analysis, the impact of CHIP mutations was HR 1.13 (0.94; 1.36) (P = .192) after adjusting for sex and tobacco consumption (Table 3). We also tested whether the number of mutations grouped as 1 mutation or 2 to 3 mutations had any impact on survival; however, both had an HR at 1.17, which was nonsignificant. We next grouped the twins according to whether they had “DTA mutations” only or “other mutations” (ie, other mutation ± DTA mutation). When adjusted for age, sex, and smoking, the negative effect of “other mutations” was not significant (HR 1.01 [0.65; 1.54], P = .98) (Table 4). In contrast, DTA mutations retained negative impact on overall survival when adjusted for age, sex, and tobacco consumption (HR 1.21 [1.01; 1.45], P = .035). Using age as the underlying time scale, DTA mutations were borderline significant (HR 1.18 [0.98; 1.41], P = .077), after adjusting for sex and tobacco consumption (Table 4).
Cox regression analysis including mutational status, sex, age, and tobacco consumption
| Variable | HR | 95% CI | P |
|---|---|---|---|
| A. | |||
| Mutation (yes/no) | 1.17 | 0.97-1.40 | .096 |
| Age, y | 1.11 | 1.08-1.13 | <.001 |
| Sex (male) | 1.15 | 0.90-1.45 | .259 |
| Smoking (1st tertile) | 1.02 | 0.81-1.29 | .840 |
| Smoking (2nd tertile) | 1.22 | 0.96-1.55 | .100 |
| Smoking (3rd tertile) | 1.71 | 1.24-2.35 | .001 |
| B. | |||
| Mutation (yes/no) | 1.13 | 0.94-1.36 | .192 |
| Sex (male) | 1.14 | 0.90-1.44 | .274 |
| Smoking (1st tertile) | 1.03 | 0.83-1.30 | .769 |
| Smoking (2nd tertile) | 1.26 | 0.99-1.59 | .052 |
| Smoking (3rd tertile) | 1.73 | 1.26-2.37 | .001 |
| Variable | HR | 95% CI | P |
|---|---|---|---|
| A. | |||
| Mutation (yes/no) | 1.17 | 0.97-1.40 | .096 |
| Age, y | 1.11 | 1.08-1.13 | <.001 |
| Sex (male) | 1.15 | 0.90-1.45 | .259 |
| Smoking (1st tertile) | 1.02 | 0.81-1.29 | .840 |
| Smoking (2nd tertile) | 1.22 | 0.96-1.55 | .100 |
| Smoking (3rd tertile) | 1.71 | 1.24-2.35 | .001 |
| B. | |||
| Mutation (yes/no) | 1.13 | 0.94-1.36 | .192 |
| Sex (male) | 1.14 | 0.90-1.44 | .274 |
| Smoking (1st tertile) | 1.03 | 0.83-1.30 | .769 |
| Smoking (2nd tertile) | 1.26 | 0.99-1.59 | .052 |
| Smoking (3rd tertile) | 1.73 | 1.26-2.37 | .001 |
In part A, time since blood sample is used as the underlying time scale, whereas age is used as the underlying the time scale in part B, emphasizing the impact of age on overall survival in this elderly cohort. The borderline association of CHIP mutations (P = .096) was not confirmed when age was used as the underlying time scale in the Cox regression analysis.
Cox regression analysis, including mutational status grouped as DTA mutation and “other mutation,” sex, age, and tobacco consumption
| Variable | HR | 95% CI | P |
|---|---|---|---|
| A. | |||
| Only DTA mutation | 1.21 | 1.01-1.45 | .035 |
| Other mutation | 1.01 | 0.65-1.55 | .981 |
| Age, y | 1.11 | 1.08-1.13 | <.001 |
| Sex (male) | 1.15 | 0.91-1.46 | .233 |
| Smoking (1st tertile) | 1.03 | 0.82-1.30 | .772 |
| Smoking (2nd tertile) | 1.22 | 0.96-1.55 | .108 |
| Smoking (3rd tertile) | 1.72 | 1.26-2.36 | .001 |
| B. | |||
| Only DTA mutation | 1.18 | 0.98-1.41 | .077 |
| Other mutation | 0.97 | 0.63-1.50 | .899 |
| Sex (male) | 1.15 | 0.91-1.44 | .247 |
| Smoking (1st tertile) | 1.04 | 0.83-1.31 | .715 |
| Smoking (2nd tertile) | 1.25 | 0.99-1.58 | .060 |
| Smoking (3rd tertile) | 1.75 | 1.28-2.38 | <.001 |
| Variable | HR | 95% CI | P |
|---|---|---|---|
| A. | |||
| Only DTA mutation | 1.21 | 1.01-1.45 | .035 |
| Other mutation | 1.01 | 0.65-1.55 | .981 |
| Age, y | 1.11 | 1.08-1.13 | <.001 |
| Sex (male) | 1.15 | 0.91-1.46 | .233 |
| Smoking (1st tertile) | 1.03 | 0.82-1.30 | .772 |
| Smoking (2nd tertile) | 1.22 | 0.96-1.55 | .108 |
| Smoking (3rd tertile) | 1.72 | 1.26-2.36 | .001 |
| B. | |||
| Only DTA mutation | 1.18 | 0.98-1.41 | .077 |
| Other mutation | 0.97 | 0.63-1.50 | .899 |
| Sex (male) | 1.15 | 0.91-1.44 | .247 |
| Smoking (1st tertile) | 1.04 | 0.83-1.31 | .715 |
| Smoking (2nd tertile) | 1.25 | 0.99-1.58 | .060 |
| Smoking (3rd tertile) | 1.75 | 1.28-2.38 | <.001 |
In part A, time since blood sample is used as the underlying time scale, whereas age is used as the underlying the time scale in part B. When we divided the mutations into DTA and “other mutations,” defined as non-DTA mutations, but with or without cooccurring DTA mutations, we found a significant association of DTA mutations after adjusting for age, sex, and tobacco consumption. This association was retained as a borderline significant association when we used age as the underlying time scale in the Cox regression.
Last, we investigated the impact of clone size on overall mortality. Subjects with mutations were categorized according to VAF <10% or ≥10%, respectively; however, no difference in overall mortality could be seen according to these cutoffs (supplemental Table 1a). Using age as the underlying time scale did not change the results notably (supplemental Table 1b).
Mortality in twins discordant for CH
In a direct comparison, we tested the association of CH with mortality after controlling for familial factors, which is a unique possibility in this twin study, where 96% of the cohort has deceased. In discordant pairs, we tested if the affected twin died before the unaffected twin. A total of 127 twin pairs were discordant for carrying a mutation, and in 61 (48%) cases, the affected twin died first (P = .72). Furthermore, we tested whether certain subtypes of CHIP mutations were directly linked to mortality by comparing discordant pairs with DTA mutated vs nonmutated (n = 104), and “other mutations” vs nonmutated (n = 23). No significant difference was observed in either group; 50 (48%) DTA-mutated twins and 11 (48%) twins with “other mutations” died before their nonmutated related twin (P = .77 and P = 1.00, respectively). Thus, these data indicate that CHIP mutations are unlikely to affect mortality in the healthy elderly, when controlling for familial factors.
Risk of hematological cancer and unspecified cytopenia
A total of 27 twins developed a hematological cancer during the follow-up, of those 8 could be categorized as myeloid neoplasms, and the remaining 19 were classified as B- or T-cell malignancies. For confidentiality reasons, we do not report on groups with <5 individuals affected; thus, the specific numbers within each hematological cancer cannot be listed. First, we tested the risk of developing a hematological cancer and its association with the presence of CHIP mutations. After adjusting for sex, age, and tobacco consumption, we did not find any association between CHIP mutations and the risk of developing a hematological cancer (HR [95% CI] 1.80 [0.87; 3.75], P = .15) (supplemental Table 2). However, in the group with unspecified cytopenia (n = 74), when we adjusted for age, sex, and tobacco consumption, we found a borderline significant association (HR [95% CI] 1.59 [0.99; 2.54], P = .052); however, CHIP mutations were the only variable with a potential association to cytopenia (supplemental Table 3).
Discussion
Here, we report the hitherto first analyses of CHIP in twins. Our primary aim was to uncover the role of genetic and familial factors for the development of CHIP. Because this twin cohort was population based and followed prospectively for 20 years, we were also able to report data on the association to hematological disease and to overall mortality. Importantly, a direct comparison of discordant twins allowed us to estimate the association between CH and mortality, controlling for familial factors.
We identified CHIP mutations in 36% of the twins, which is in line with what is reported in other studies of this age group,8,10 and, as expected, we mainly identified DTA mutations. It is still not clear why DTA genes are so frequently mutated during aging, and it has been speculated whether this may be caused by a genetic predisposition.19,20 However, we did not find any indication of a genetic predisposition to CH in the current study. This is in contrast to what was suggested by 2 previous studies, which detected an aggregation of TET2 mutations in siblings20 and was observed as a possible germline defect that may associate with CH with or without driver mutations.19 Our data were cleaned for polymorphisms and rare variants with a VAF of 40% to 60%, so these data did not bias the results on genetic predisposition. Using this strategy, we report only 4 mutations with a VAF between 40% and 60%, which were 2 truncating TET2 mutations and 2 JAK2 V617F mutations.
We next tested different aspects of CH, such as the impact of individual mutations and allele frequency, but none of these pointed to a genetic predisposition; in particular, we could not confirm a predisposition to TET2 mutations as previously suggested.20 Jaiswal et al have shown a significant impact of CHIP mutations on overall mortality, with an HR of 1.4 on all-cause mortality after adjusting for both age and sex.5 By contrast, we did not find CHIP mutations to have any significant impact on overall mortality in the elderly population in this study where age was used as the underlying time scale in the survival analysis. DTA mutations did retain a borderline significant impact using age as underlying time scale (P = .077) in contrast to “other mutations,” which did not have any impact on overall mortality, even though this group included patients with >1 mutation and mutations in DNA repair genes. Meanwhile, our findings are in line with the study from van den Akker et al, who applied a left truncated Cox proportional hazard model and showed that CHIP mutations were not associated with overall mortality in the elderly population.33 In the current study, we had the unique possibility of investigating the impact of CHIP when controlling for familial factors (ie, by comparing discordant twin pairs). Interestingly, we identified no significant difference between affected and unaffected discordant twins. Thus, based on the current data, CHIP mutations per se are not associated with increased mortality. However, it is important to acknowledge the differences in population age, when comparing studies, because our study population is older than those reported in most of the previous studies investigating the impact of CHIP on survival.4,5 CHIP might have an impact on survival if present in younger individuals, but age-dependent incidents, such as CHIP, can lose their negative impact in the elderly.
In addition, we did not observe any association with the development of hematological cancers; however, there was a tendency (P = .052) that twins with CHIP mutations had an increased risk of developing “unspecified cytopenia.” It can be speculated whether this relates to the development of clonal cytopenia of undetermined significance9 ; however, the MDS diagnostic tools and criteria have also been refined and optimized significantly over the past 2 decades, so it is likely that MDS has been underdiagnosed in this study, where cases are registered from 1997 and onward. The association between CHIP and “unspecified cytopenia” needs to be validated in other cohorts, because the definition of “unspecified cytopenia” is not consistent.
In the current study, we did not find any aggregation of hematological cancer or unspecified cytopenia within twin pairs. This being said, it is becoming increasingly clear that some myeloid neoplasms are caused by a genetic predisposition.34 Some hereditary myeloid cancers present late in life, without any obvious predisposition syndromes and with variable penetrance.35 However, we were not able to detect any genetic predisposition to CH or TET2 mutations, specifically, in this population-based cohort of elderly twins.
Limitations
There were no complete blood counts performed at the time of blood collection in our study, so it was not possible to assess the impact and correlation with peripheral blood counts. However, this does not influence the main purpose with the study, which was to investigate the possible genetic predisposition to CH.
In this study, we only examined elderly twins over the age of 73 years, where CH is a common phenomenon. By only investigating elderly twins, we may theoretically have missed a genetic predisposition to CH appearing in younger individuals. However, investigating younger twins will require a larger cohort, because the proportion with CHIP mutations would be much lower.
In conclusion, this is hitherto the first twin study investigating a genetic predisposition to CH and the impact of CH on mortality in twins. In this cohort, no germline predisposition to CHIP mutations could be detected. Furthermore, no difference in mortality was observed in a direct comparison among twin pairs that were discordant for CHIP mutations.
Please contact the corresponding author for questions regarding the original data.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work is supported by a grant from the Danish Cancer Society, grant R204-A12363, and center grants from the Danish Cancer Society, Danish Research Center for Precision Medicine in Blood Cancer, grant 223-A13071-18-S68 (K.G., J.W.H.), and the Novo Nordisk Foundation, Novo Nordisk Foundation Center for Stem Cell Biology, DanStem; grant NNF17CC0027852 (K.G., J.W.H.), and the Greater Copenhagen Health Science Partners (Clinical Academic Group in Translational Hematology). This work is also supported by the Odense University Hospital AgeCare program (Academy of Geriatric Cancer Research) (D.A.P., L.A.L., S.B.C., J.H., K.C.). The Danish Aging Research Center is supported by a grant from the VELUX Foundation, grant Velux 31205. The Danish Twin Registry has been supported by The National Program for Research Infrastructure 2007 grant 09-063256 from the Danish Agency for Science Technology and Innovation, the Velux Foundation, and the National Institutes of Health, National Institute on Aging grant P01 AG08761.
Authorship
Contribution: J.W.H., K.C., and K.G. conceived the study and wrote the manuscript; J.W.H., S.H., F.F., and J.W. did the sequencing work and analyzed the sequencing data; D.A.P., L.A.L., S.B.C., and J.H. linked the data and performed the twin and survival analyses; and all authors contributed to the manuscript and interpretation of the data and approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Kirsten Grønbæk, BRIC, Ole Maaløesvej 5, Building 2, 3rd Floor, Section 3733, 2200 Copenhagen, Denmark; e-mail: kirsten.groenbaek@regionh.dk.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal